Medicare Drug Price Negotiation Program and Medicare Prescription Drug Benefit Program
This proposed rule would codify the Medicare Drug Price Negotiation Program ("Negotiation Program") and would establish certain new policies for the Negotiation Program and the ...
Centers for Medicare & Medicaid Services (CMS), Health and Human Services (HHS).
ACTION:
Proposed rule.
SUMMARY:
This proposed rule would codify the Medicare Drug Price Negotiation Program (“Negotiation Program”) and would establish certain new policies for the Negotiation Program and the Medicare Prescription Drug Benefit Program as required by the Inflation Reduction Act of 2022. This proposed rule would also propose a modification to the fixed combination drug policy.
DATES:
To be assured consideration, comments must be received at one of the addresses provided below, no later than 5 p.m. on August 17, 2026.
ADDRESSES:
In commenting, please refer to file code CMS-4215-P.
Comments, including mass comment submissions, must be submitted in one of the following three ways (please choose only one of the ways listed):
2.
By regular mail.
You may mail written comments to the following address ONLY: Centers for Medicare & Medicaid Services, Department of Health and Human Services, Attention: CMS-4215-P, P.O. Box 8013, Baltimore, MD 21244-8013.
Please allow sufficient time for mailed comments to be received before the close of the comment period.
3.
By express or overnight mail.
You may send written comments to the following address ONLY: Centers for Medicare & Medicaid Services, Department of Health and Human Services, Attention: CMS-4215-P, Mail Stop C4-26-05, 7500 Security Boulevard, Baltimore, MD 21244-1850.
For information on viewing public comments, see the beginning of the
SUPPLEMENTARY INFORMATION
section.
Inspection of Public Comments:
All comments received before the close of the comment period are available for viewing by the public, including any personally identifiable or confidential business information that is included in a comment. We post all comments received before the close of the comment period on the following website as soon as possible after they have been received:
https://www.regulations.gov.
Follow the search instructions on that website to view public comments. CMS will not post on
Regulations.gov
public comments that make threats to individuals or institutions or suggest that the commenter will take actions to harm an individual. CMS continues to encourage individuals not to submit duplicative comments. We will post acceptable comments from multiple unique commenters even if the content is identical or nearly identical to other comments.
This proposed rule would codify policies related to the implementation of certain provisions of the Inflation Reduction Act of 2022 (IRA) (Pub. L. 117-169, August 16, 2022) and amendments made by the Working Families Tax Cut legislation (Pub. L. 119-21, July 4, 2025).
This proposed rule would also codify policies for the Medicare Drug Price Negotiation Program at part 429 consistent with sections 1191 through 1198 of the Social Security Act (hereinafter “the Act”) and codify policies for the Medicare Prescription Drug Benefit Program at part 423 consistent with section 11001(b) of the IRA, which made certain amendments to the Act, including with respect to Medicare Part D.
2. Summary of the Provisions
We propose to codify policies established in final guidance for the Negotiation Program [1]
in regulatory text. Specifically, we propose to codify, with limited modification, the policies set forth in guidance for the Medicare Drug Price Negotiation Program by adding the new part 429 to title 42, Chapter IV of the Code of Federal Regulations and modifying policies for the Medicare Prescription Drug Benefit Program at part 423 and welcome comments on these proposals.
In addition, we propose new policies for the Medicare Drug Price Negotiation Program as follows:
Proposed § 429.125(b)(4)(i) would clarify treatment of new formulations were circumstances to emerge where statutory requirements could be in tension with the general fixed combination drug policy proposed at § 429.125(b)(4). To do so, we are proposing a narrow modification to the general fixed combination drug policy for certain fixed combination drugs that are new formulations. Under the modification, if CMS determines that products with the same New Drug Application (NDA)/Biologics License Application (BLA) holder differ in active moiety(ies)/active ingredient(s) due to the inclusion of an active moiety/active ingredient that creates a new formulation and enables an alternative route of administration for the co-administered active moiety(ies)/active ingredient(s), then CMS will identify the potential qualifying single source drug using all dosage forms and strengths of the shared active moiety(ies)/active ingredient(s) that is offered by the same NDA/BLA holder.
Proposed § 429.125(c)(3)(i) would clarify how CMS would identify the day from which to measure the 7- and 11-year time since approval and licensure periods for drugs that formerly qualified for the Orphan Drug Exclusion.
Proposed § 429.130 would codify the process and schedule according to which CMS reviews information to determine if the manufacturer of a generic drug or biosimilar that is approved or licensed, respectively, is engaged in Bona Fide Marketing (as defined in § 429.20) of that generic drug or biosimilar.
Proposed § 429.210(c) would provide additional details related to the Primary Manufacturer transfer of responsibility for all requirements of the Negotiation Program Agreement to an acquiring entity.
( printed page 36237)
Proposed § 429.415(a)(2) would explain how CMS would calculate the 30-day equivalent supply for a selected drug that is typically administered one time (for example, some vaccines, gene therapies, and cancer therapies).
Proposed § 429.440 would explain how CMS would implement the Temporary Floor for Small Biotech Drugs for initial price applicability years 2029 and 2030.
Proposed §§ 429.605 and 429.610 would clarify when off-label use would be considered for renegotiation eligibility and selection by aligning the renegotiation eligibility and selection policies for off-label use with the initial offer development process. This clarification maintains consistency across CMS processes for negotiation and renegotiation, as required by section 1194(f)(4)(B) of the Act.
Unless otherwise specified, CMS proposes that the provisions herein would apply with respect to all initial price applicability years beginning with initial price applicability year 2029, including, for example, with respect to the selection of drugs and the negotiation or renegotiation of MFPs for initial price applicability year 2029 that will take place during calendar year 2027. In this proposed rule, unless otherwise specified, references hereinafter to “the Negotiation Program Guidance” are to the most recent program guidance published by CMS, which is the Medicare Drug Price Negotiation Program: Final Guidance, Implementation of Sections 1191-1198 of the Act for Initial Price Applicability Year 2028 and Manufacturer Effectuation of the Maximum Fair Price in 2026, 2027, and 2028 that was published on September 30, 2025.
CMS anticipates publishing the final version of this rule in Fall 2026, after considering and responding to public comments received on this proposed rule, such that the final requirements will apply to initial price applicability year 2029 and all subsequent years, taking effect beginning with the process of the selection of drugs for negotiation and renegotiation, if applicable, for initial price applicability year 2029.
Given that the identification and selection of drugs for negotiation and renegotiation occurs more than 2 years before the first application of the MFP (that is, before the start of the selected drug's first initial price applicability year), the processes for drugs that were selected for negotiation for initial price applicability years 2026, 2027, and 2028 and for renegotiation for initial price applicability year 2028 will be at varying stages of implementation when this rule is proposed and finalized. Consistent with the program instruction requirement at sections 11001(c) and 11002(c) of the IRA, the program guidance issued by CMS for initial price applicability years 2026, 2027, and 2028 remains applicable and is not superseded by this proposed rule with respect to such years. In other words, because sections 11001(c) and 11002(c) of the IRA require CMS to implement the Negotiation Program for initial price applicability years 2026, 2027, and 2028 through program instruction and other forms of program guidance, the requirements for a selected drug that was included on the list of selected drugs with respect to initial price applicability year 2026, 2027, or 2028 are set forth with respect to such years in the applicable program guidance. Revisions to the implementation of policy for 2026, 2027, and 2028 with respect to drugs selected for initial price applicability years 2026, 2027, and 2028 would be addressed by CMS through publication of revised guidance. In accordance with the expiration of the statutory program instruction requirement at the end of 2028, CMS proposes that the provisions herein, as applicable, will apply starting in 2029 with respect to the drugs selected for initial price applicability years of 2026, 2027, or 2028.
Finally, CMS reminds interested parties that the exclusion for small biotech drugs from what is otherwise a negotiation-eligible drug under section 1192(d)(2) of the Act ended in initial price applicability year 2028 and is, therefore, not codified in this rule. However, the definition of an eligible small biotech drug for purposes of the calculation of the temporary floor on the maximum fair price for small biotech drugs under section 1194(d) of the Act is included in this proposed rule (as described in more detail in section II.E.2. of this proposed rule).
3. Summary of Costs and Benefits
We are proposing new policies for the Negotiation Program as follows: a modification to the general fixed combination drug policy to clarify our treatment of certain new formulations; clarification for drugs that formerly qualified for the orphan drug exclusion; revisions regarding process and schedule of CMS review of information in making the determination for Bona Fide Marketing; additional details related to the Primary Manufacturer transfer of responsibility for all requirements of the Negotiation Program Agreement to an acquiring entity; calculation of the 30-day equivalent supply for a selected drug that is typically administered one time; implementation of the Temporary Floor for Small Biotech Drugs for initial price applicability years 2029 and 2030; and clarification of off-label use in consideration for renegotiation eligibility and selection. In summary, the effects of the IRA are to reduce government expenditures for Part B, to increase expenditures for Part D through 2030, and to decrease Part D expenditures beginning in 2031. For a detailed discussion of the economic impacts, see section V. of this proposed rule.
B. Background
Sections 11001 and 11002 of the Inflation Reduction Act of 2022 (IRA) (Pub. L. 117-169), signed into law on August 16, 2022, establish the Medicare Drug Price Negotiation Program (hereinafter the “Negotiation Program”) to negotiate maximum fair prices (MFPs) for certain high expenditure, single source drugs and biological products. Specifically, in accordance with section 1191(c)(3) of the Act, MFP means, with respect to a year during a price applicability period and with respect to a selected drug (as defined in section 1192(c) of the Act) with respect to such period, the price negotiated pursuant to section 1194 of the Act, and updated pursuant to section 1195(b) of the Act, as applicable, for such drug and year. The requirements for this program are described in sections 1191 through 1198 of the Act, as added by sections 11001 and 11002 of the IRA and as amended by section 71203 of the “Working Families Tax Cut” legislation (Pub. L. 119-21).
Under the IRA, with respect to each initial price applicability year, CMS shall: (1) publish a list of selected drugs in accordance with section 1192 of the Act; (2) enter into agreements with manufacturers of selected drugs in accordance with section 1193 of the Act; (3) negotiate MFPs for such selected drugs in accordance with section 1194 of the Act; (4) publish MFPs for selected drugs in accordance with section 1195 of the Act; (5) carry out administrative duties and compliance monitoring in accordance with section 1196 of the Act; and (6) impose civil monetary penalties (CMPs) in accordance with section 1197 of the Act. With respect to initial price applicability year 2028 and subsequent years, in accordance with section 1194(f) of the Act, CMS shall also: (1) determine renegotiation-eligible drugs; (2) determine whether to select drugs for renegotiation; and (3) renegotiate the MFP for any drug selected for renegotiation. To the extent applicable, any references in this proposed rule to the “MFP” include a renegotiated MFP.
( printed page 36238)
Section 1198 of the Act establishes certain limitations on administrative and judicial review relevant to the Negotiation Program.
Additionally, on July 4, 2025, the “Working Families Tax Cut” legislation was signed into law. Section 71203(a)(2) of the “Working Families Tax Cut” legislation amended section 1192(e)(1)(3)(A) of the Act to modify the requirements for a drug to qualify for the Orphan Drug Exclusion. Section 71203(a)(3) of the “Working Families Tax Cut” legislation also added new section 1192(e)(4) of the Act, which describes the treatment of former orphan drugs.
For the first year of the Negotiation Program, the Secretary of the U.S. Department of Health and Human Services (“the Secretary”) selected 10 high expenditure, single source drugs covered under Part D for negotiation. The negotiated MFPs for these drugs took effect in initial price applicability year 2026.[2]
The Secretary selected an additional 15 drugs covered under Part D for negotiation for initial price applicability year 2027,[3]
and 15 drugs covered under Part D and/or payable under Part B for initial price applicability year 2028.[4]
The Secretary will select up to 20 drugs covered under Part D and/or payable under Part B for initial price applicability year 2029 and subsequent initial price applicability years. Beginning with initial price applicability year 2028, the Secretary could also select drugs from initial price applicability year 2026 and subsequent initial price applicability years for renegotiation. The Secretary selected one drug for renegotiation for initial price applicability year 2028.[5]
For initial price applicability years 2026 through 2028 of the Negotiation Program, sections 11001(c) and 11002(c) of the IRA direct CMS to implement the Negotiation Program through program instruction and other forms of program guidance. CMS issued initial or draft versions of program guidance for each initial price applicability year 2026,[6]
2027,[7]
and 2028 [8]
and requested public comment on each version. CMS then issued a revised or final version of the guidance for each of these program years.[9 10 11]
This proposed rule proposes to codify these requirements at parts 423 and 429 to title 42, chapter IV of the Code of Federal Regulations to implement sections 11001 and 11002 of the IRA. The effective date of the proposed provisions are discussed in section I.A.2. of this proposed rule.
Consistent with the program instruction requirement of sections 11001(c) and 11002(c) of the IRA, CMS will issue program guidance related to manufacturer effectuation of the MFP for 2028, including with respect to drugs payable under Part B. CMS intends to codify requirements related to MFP effectuation for 2029 and subsequent years in future rulemaking. CMS stated in the Negotiation Program Guidance its intent to codify MFP effectuation policies for 2029 and subsequent years after guidance for 2028 has been finalized.
C. Severability of Provisions
Finally, CMS is clarifying and emphasizing its intent that if any provision of this rule, once finalized, is held to be invalid or unenforceable by its terms, or as applied to any person or circumstance, or stayed pending further agency action, it shall be severable from this rule and not affect the remainder thereof or the application of the provision to other persons not similarly situated or to other, dissimilar circumstances. Through this rule, CMS proposes provisions that are intended to and will operate independently of each other, even if each serves the same general purpose or policy goal. Where a provision is necessarily dependent on another, the context generally makes that clear (such as by a cross-reference to apply the same standards or requirements).
II. Proposed Requirements for the Medicare Drug Price Negotiation Program
A. General Provisions
1. Basis and Scope (§ 429.10)
In proposed § 429.10, we would state that part 429 implements sections 1191 through 1198 of the Act and sections 11001 and 11002 of the IRA, which set forth the requirements of the Medicare Drug Price Negotiation Program. The Medicare Drug Price Negotiation Program requires the Secretary to negotiate and renegotiate, for applicable periods, Medicare prices for certain high expenditure, single source drugs and biological products.
Additionally, in proposed § 429.10(c), we would state that, were any provision of part 429 to be held invalid or unenforceable by its terms, or as applied to any person or circumstance, such provision would be severable from part 429 and the invalidity or unenforceability would not affect the remainder thereof or any other part of this subchapter or the application of such provision to other persons not similarly situated or to other, dissimilar circumstances. While the provisions in part 429 are intended to present a comprehensive approach to implementing the Medicare Drug Price Negotiation Program, we intend that each of them is a distinct, severable provision, as proposed. Through this rulemaking, the proposed policies contained herein are intended to operate independently of each other, even if each serves the same general purpose or policy goal. For example, we intend that the proposed policies related to requests for a biosimilar delay (proposed § 429.110) are distinct and severable from the proposals related to the identification of qualifying single source drugs (proposed § 429.125). As another example, we intend that the proposed policy for additional price exchange opportunities for purposes of providing additional flexibility to extend and consider offers and counteroffers (proposed § 429.530) is distinct and severable from the proposals for CMS and Primary Manufacturers (as defined in proposed § 429.20) to submit written initial offers and statutory written counteroffers, respectively, for purposes of determining an agreed-upon maximum fair price (MFP) (proposed § 429.520(a) and 429.525(a), respectively). Even where one provision makes reference to a second provision, § 429.10(c) clarifies the intent of the agency is that the two provisions would be severable if one provision were to be invalidated in whole or in part. For
( printed page 36239)
example, we would still be able to adjust the preliminary price based on section 1194(e)(1) factors (as defined in proposed § 429.20) as described in proposed § 429.510(f) even if the provision for Primary Manufacturers to submit market data and revenue and sales volume data for the selected drug in the United States is deemed invalid (proposed § 429.505(b)(2)(v)).
2. Definitions (§ 429.20)
In this proposed rule, we would codify the definitions of terms consistent with the meanings given in sections 1191 through 1198 of the Act or established in the Negotiation Program Guidance, as applicable, as well as proposing to codify new definitions based on policies detailed in this proposed rule.
a. Additional Delay Request
We propose to define “Additional Delay Request” as a request to delay the inclusion on the selected drug list of a reference drug for which an initial delay request has been granted for a second initial price applicability year consistent with proposed § 429.110 and section 1192(f)(1)(B)(i)(II) of the Act.
b. Applicable Program Agreement
We propose to define “applicable program agreement” as an agreement under the Manufacturer Discount Program as specified in section 1860D-14C of the Act or a rebate agreement described in section 1927(b) of the Act.
c. Authorized Generic Drug
We propose to define “authorized generic drug” in accordance with the definition of such term at section 1192(e)(2)(B) of the Act. Section 1192(e)(2)(B)(i) of the Act defines an “authorized generic drug” that is a drug as a drug as defined in section 505(t)(3) of the Federal Food, Drug, and Cosmetic (FD&C) Act. Section 1192(e)(2)(B)(ii) of the Act defines an authorized generic drug that is a biological product as a product that has been licensed under section 351(a) of the Public Health Service (PHS) Act and is marketed, sold, or distributed directly or indirectly to the retail class of trade under a different labeling, packaging (other than repackaging as the reference product in blister packs, unit doses, or similar packaging for institutions), product code, labeler code, trade name, or trademark than the reference product.
d. Authorized Representative
We propose to define “authorized representative” as an individual that has the authority or capacity to legally bind the Primary Manufacturer to the terms and conditions of the Negotiation Program Agreement and meets one of the following criteria:
Chief Executive Officer of the Primary Manufacturer.
Chief Financial Officer of the Primary Manufacturer.
An individual with equivalent authority to a Chief Executive Officer or Chief Financial Officer of the Primary Manufacturer.
An individual that has been granted delegation of signature authority on behalf of one of the individuals specified in paragraphs (1) through (3) of this proposed definition.
We solicit comment on this proposed definition and potential alternative formulations, including whether to adopt a broader definition to account for other contexts within the Negotiation Program, such as an individual making a submission to CMS on behalf of an entity other than a Primary Manufacturer.
e. Average Manufacturer Price (AMP)
We propose to define “average manufacturer price (AMP)” as having the meaning given such term in section 1927(k)(1) of the Act.
f. Average Non-Federal Average Manufacturer Price (non-FAMP)
We propose to define “average non-Federal Average Manufacturer Price (non-FAMP)” as having the meaning set forth in section 1194(c)(6) of the Act.
g. Average Sales Price (ASP)
We propose to define “average sales price (ASP)” as the manufacturer's price for a quarter for a drug represented by a particular 11-digit National Drug Code (NDC-11) determined under § 414.804 and as reported in section 1927(b)(3) of the Act.
h. Billing Unit
We propose to define “billing unit” as the identifiable quantity of a drug or biological product associated with a billing and payment code (for example, a Healthcare Common Procedure Coding System code), as established by CMS.
i. Biologics Licenses Application (BLA)
We propose to define “Biologics License Application (BLA)” as an application submitted under section 351 of the PHS Act.
j. Biosimilar Biological Product or Biosimilar
We propose to define “biosimilar biological product” or “biosimilar” as having the meaning given such term in section 1847A(c)(6) of the Act. For purposes of these regulations, we use the terms “biosimilar biological product” and “biosimilar” interchangeably when describing the requirements of sections 11001 and 11002 of the IRA. Specifically, section 1192(f)(5) of the Act, as added by section 11002 of the IRA, uses the meaning given to “biosimilar biological product” from section 1847A(c)(6) of the Act. Proposed part 429 uses the term “biosimilar” unless otherwise specified, such as related to the “Biosimilar” included in a Biosimilar Delay Request under section 11002 of the IRA in proposed § 429.20.
k. Biosimilar Delay Request
We propose to define “Biosimilar Delay Request” as either an Initial Delay Request or an Additional Delay Request.
l. Biosimilar Manufacturer
We propose to define “Biosimilar Manufacturer” as one of the following:
The BLA holder for the Biosimilar.
If a BLA has been submitted to the U.S. Food and Drug Administration (FDA) for review but the Biosimilar has not been licensed, the sponsor of the BLA submitted for review by the FDA.
If the Biosimilar has not been licensed and the BLA has not been submitted to the FDA, the organization planning to be the sponsor when the BLA is submitted for review by the FDA.
We believe that this approach is appropriate because: (1) it clearly identifies one manufacturer that may submit a Biosimilar Delay Request for a given Biosimilar, avoiding the possibility that we would receive two such requests naming the same Biosimilar for the same initial price applicability year; and (2) the status of the application for licensure for the Biosimilar is material to CMS' consideration of a request for a Biosimilar Delay Request, as described in section II.B.3. of this proposed rule.
m. BLA Holder
We propose to define “BLA holder” as the entity that is the holder of the license(s) permitting marketing of a biological product in accordance with section 351 of the PHS Act.
n. Bona Fide Marketing
We propose to define “Bona Fide Marketing” as having the meaning set forth in proposed § 429.130(a). Further discussion of CMS' review of one or more manufacturers of an approved generic drug or licensed biosimilar engaging in Bona Fide Marketing is included in section II.B.6.d. of this proposed rule.
( printed page 36240)
o. Combined Part B and Part D Amount
We propose to define “combined Part B and Part D amount” as an amount equal to the weighted average of the payment amount under section 1847A(b)(4) of the Act and the sum of the plan-specific enrollment weighted amount as determined by CMS under proposed § 429.420(c).
p. CPI-U
We propose to define “CPI-U” as the monthly Consumer Price Index for All Urban Consumers (United States city average) index level for all items from the Bureau of Labor Statistics.
q. Direct and Indirect Remuneration (DIR)
We propose to define “Direct and Indirect Remuneration (DIR)” as having the meaning set forth in 42 CFR 423.308.
r. Drug Covered Under Part D
We propose to define “drug covered under Part D” as a covered part D drug as defined in section 1860D-2(e) of the Act. We acknowledge that section 1860D-2(e) of the Act defines the term “covered part D drug” rather than “drug covered under Part D”. For purposes of this rule, we use the term “drug covered under Part D” for simplicity and intend this term to be synonymous with the statutory term “covered part D drug”.
s. Drug Payable Under Part B
We propose to define “drug payable under Part B” as a drug or biological product for which payment may be made under part B of Title XVIII of the Act.
t. Estimated Remuneration at Point-of-Sale Amounts (ERPOSA)
We propose to define “estimated renumeration at point-of-sale amounts (ERPOSA)” as the estimated amount of rebates or other price concessions that the Part D plan sponsor is required to apply, or has elected to apply, to the negotiated price as a reduction in the drug price made available to the beneficiary at the point of sale.
u. Extended-Monopoly Drug
We propose to define “extended-monopoly drug” as having the meaning set forth in section 1194(c)(4) of the Act.
v. FDA-Approved Indication
We propose to define “FDA-approved indication” as the information included in drug labeling per 21 CFR 201.57(c)(2) or FDA regulation(s) as applicable.
w. Fixed Combination Drug
We propose to define “fixed combination drug” as having the meaning set forth in 21 CFR 300.50.
x. Generic Drug
We propose to define “generic drug” as a drug approved in an Abbreviated New Drug Application (ANDA) under section 505(j) of the Federal Food, Drug, and Cosmetic Act (“FD&C Act”).
y. Healthcare Common Procedure Coding System (HCPCS) Code
We propose to define “Healthcare Common Procedure Coding System (HCPCS) code” as a billing and payment code, as established by CMS for payment under Part B, used to describe a drug or biological and for which CMS may publish a payment amount.
z. High Likelihood Deadline
We propose to define “High Likelihood Deadline” as the date that is 2 years after the statutorily defined selected drug publication date for the initial price applicability year for which the reference drug would be included on the selected drug list absent a successful Initial Delay Request. This period of time is consistent with time periods specified in sections 1192(f)(1)(A) and (2)(B)(i)(I) of the Act.
aa. Initial Delay Period
We propose to define “Initial Delay Period” as the time period between: (1) the selected drug publication date for the initial price applicability year for which the Reference Drug otherwise would have been included on the selected drug list but for the successful Initial Delay Request, as proposed in § 429.110(g); and (2) the selected drug publication date with respect to the initial price applicability year that is 1 year after the initial price applicability year for which the Reference Drug otherwise would have been included on the selected drug list but for the successful Initial Delay Request as set forth in section 1192(f)(2) of the Act.
ab. Initial Delay Request
We propose to define “Initial Delay Request” as a request to delay the inclusion of a reference drug on the selected drug list by one initial price applicability year consistent with proposed § 429.110(c) and section 1192(f)(1)(B)(i)(I) of the Act.
ac. Initial Price Applicability Year
We propose to define “initial price applicability year” as having the meaning set forth in section 1191(b)(1) of the Act.
ad. Knowingly
We propose to define “knowingly” as having the meaning set forth in 42 CFR 1003.110.
ae. Long-Monopoly Drug
We propose to define “long-monopoly drug” as having the meaning set forth in section 1194(c)(5) of the Act.
af. Manufacturer
We propose to define “manufacturer” as having the meaning set forth in section 1191(c)(1) of the Act.
ag. Manufacturer Discount Program
We propose to define “Manufacturer Discount Program” to mean the Medicare Part D Manufacturer Discount Program established under section 1860D-14C of the Act.
ah. Maximum Fair Price (MFP)
We propose to define “maximum fair price (MFP)” as having the meaning set forth in section 1191(c)(3) of the Act.
ai. Medicare Drug Price Negotiation Program (or Negotiation Program)
We propose to define “Medicare Drug Price Negotiation Program (or Negotiation Program)” as the program created by sections 11001 and 11002 of the Inflation Reduction Act and codified in sections 1191 through 1198 of the Act and as amended.
aj. Medicare Drug Price Negotiation Program Agreement (or Negotiation Program Agreement)
We propose to define “Medicare Drug Price Negotiation Program Agreement (or Negotiation Program Agreement)” as the agreement between a Primary Manufacturer and CMS as set forth in proposed § 429.200 of this chapter and section 1193(a) of the Act.
ak. NDA Holder
We propose to define “NDA holder” as the entity that is the holder of the approval(s) to market a drug product in accordance with section 505(c) of the FD&C Act.
al. Negotiation-Eligible Drug
We propose to define “negotiation-eligible drug” as having the meaning set forth in section 1192(d) of the Act. We refer readers to proposed § 429.115 (and related discussion in section II.B.4. of this proposed rule) for CMS' proposals for identifying drugs that meet this statutory definition.
am. Negotiation Period
We propose to define “negotiation period” as having the meaning set forth in section 1191(b)(4) of the Act.
( printed page 36241)
an. Net Part D Plan Payment and Beneficiary Liability
We propose to define “Net Part D Plan Payment and Beneficiary Liability” as, for purposes of the Medicare Drug Price Negotiation Program, the total gross covered prescription drug cost for a selected drug covered under Part D net of direct and indirect remuneration (DIR) and Manufacturer Discount Program payments and excluding prescription drug event (PDE) records for which a compound code indicates the PDE record is for a compounded drug.
ao. New Drug Application (NDA)
We propose to define “New Drug Application (NDA)” as an application submitted under section 505(b) of the FD&C Act.
ap. Off-Label Use
We propose to define “off-label use” as the use for a condition for a selected drug or therapeutic alternative that is not an FDA-approved indication but is included in evidence-based clinical practice guidelines and is a medically accepted indication payable under Part B or covered under Part D or both, taking into consideration major drug compendia, authoritative medical literature, and accepted standards of medical practice, or some combination thereof.
aq. Orphan Drug Designation
We propose to define “orphan drug designation” as the meaning set forth in 21 CFR 316.3(b)(11).
ar. Outcomes
We propose to define “outcomes” as the impact of an intervention, which may be clinical or related to the functioning, symptoms, quality of life, or other aspects of a patient's life.
as. Part B Data
We propose to define “Part B data” as having the meaning of Original Medicare (OM) Part B claims data and Medicare Advantage (MA) encounter data for Part B items or services.
at. Partnership
We propose to define “partnership” as having the meaning set forth in section 1192(f)(1)(C)(ii) of the Act.
au. Personally Identifiable Information (PII)
We propose to define “personally identifiable information (PII)” as having the meaning set forth at 2 CFR 200.1.
av. Plasma-Derived Product
We propose to define “plasma-derived product” as having the meaning set forth in section 1192(e)(3)(C) of the Act.
aw. Preliminary Price
We propose to define “preliminary price” as the numerical dollar amount used by CMS in developing an initial offer in accordance with § 429.510(e) by adjusting the starting point of a selected drug based on section 1194(e)(2) factors.
ax. Price Applicability Period
We propose to define “price applicability period” as having the meaning set forth in section 1191(b)(2) of the Act.
ay. Primary Manufacturer
We propose to define “Primary Manufacturer” as the manufacturer identified by CMS as the NDA holder or the BLA holder for the selected drug.
az. Private Label Distributor
We propose to define “private label distributor” as having the meaning set forth in 21 CFR 207.1.
ba. Protected Health Information (PHI)
We propose to define “protected health information (PHI)” as having the meaning set forth at 45 CFR 160.103.
bb. Qualifying Single Source Drug
We propose to define “qualifying single source drug” as having the meaning set forth in section 1192(e) of the Act. We refer readers to proposed § 429.125 (and related discussion in section II.B.6. of this proposed rule) for CMS' proposals for identifying drugs that meet this statutory definition.
bc. Rare Disease or Condition
Section 1192(e)(3)(A) of the Act describes “rare disease or condition” as having the definition used for such term in section 526(a)(2) of the FD&C Act. Therefore, we propose to define “rare disease or condition” as having the meaning set forth in section 526(a)(2) of the FD&C Act.
bd. Reference Drug
We propose to define “Reference Drug” as a negotiation-eligible drug that includes the reference product for the biosimilar as described in section 1192(f)(1)(B) of the Act.
be. Reference Manufacturer
We propose to define “Reference Manufacturer” as the Primary Manufacturer of the Reference Drug that is named in a Biosimilar Delay Request.
bf. Reference Product
We propose to define “Reference Product” as having the meaning set forth in section 1191(c)(4) of the Act.
bg. Relabeler
We propose to define “relabeler” as having the meaning set forth in 21 CFR 207.1.
bh. Renegotiation-Eligible Drug
We propose to define “renegotiation-eligible drug” as having the meaning set forth in section 1194(f)(2) of the Act.
bi. Repackager
We propose to define “repackager” as having the meaning given the term “repacker” set forth in 21 CFR 207.1.
bj. Request To Terminate
We propose to define “Request to Terminate” as a written request submitted by a Primary Manufacturer to CMS, that CMS determines meets the conditions described in § 429.205(b)(1)(A) and (B), to request termination of its applicable program agreements in the context of a Primary Manufacturer's decision not to enter into or to terminate a Negotiation Program Agreement.
bk. Secondary Manufacturer
We propose to define “Secondary Manufacturer” as a manufacturer of a drug product included in the selected drug, that is not the Primary Manufacturer for the selected drug, and that either: (1) is listed as a manufacturer in an NDA or BLA for the selected drug; or (2) markets the selected drug pursuant to an agreement with the Primary Manufacturer but is not listed on an NDA or BLA of the selected drug. A Secondary Manufacturer includes any manufacturer of any authorized generic drug(s) and any repackager or relabeler of the selected drug that meet either of these criteria.
bl. Second Delay Period
We propose to define “Second Delay Period” as the time period between (1) the publication date of the selected drug list for initial price applicability year that is 1 year after the initial price applicability year for which the Reference Drug would have been included on the selected drug list but for the successful Initial Delay Request and (2) the publication date of the selected drug list for initial price applicability year that is 2 years after the initial price applicability year for which the Reference Drug would have been included on the selected drug list but for the successful Initial Delay
( printed page 36242)
Request as set forth in section 1192(f)(2) of the Act.
bm. Section 1194(e)(1) Factors
We propose to define “section 1194(e)(1) factors” as the factors described in section 1194(e)(1) of the Act.
bn. Section 1194(e)(2) Factors
We propose to define “section 1194(e)(2) factors” as the factors described in section 1194(e)(2) of the Act.
bo. Selected Drug
We propose to define “selected drug” as having the meaning set forth in section 1192(c) of the Act. We refer readers to proposed § 429.105 (and related discussion in section II.B.2. of this proposed rule) for CMS' proposals for identifying drugs that meet this statutory definition.
bp. Selected Drug Publication Date
We propose to define “selected drug publication date” as having the meaning set forth in section 1191(b)(3) of the Act.
bq. Self-Administered Drug
We propose to define “self-administered drug” to mean, a drug or biological that is identified by the U.S. Department of Health and Human Services Office of Inspector General (OIG) as a self-administered drug pursuant to section 1847A(g)(1) of the Act.
br. Sequestration Payment Adjustment
We propose to define “sequestration payment adjustment” to mean, when applicable, the amount that is applied to a Part B claim to determine the Medicare payment amount—after determining coinsurance, deductible, merit-based incentive payment adjustments, and any applicable Medicare Secondary Payment adjustments.
bs. Small Biotech Drug
We propose to define “Small Biotech Drug” as meaning a drug that is determined by CMS under the proposed § 429.440(b)(2), in accordance with section 1192(d)(2) of the Act, as eligible for the Temporary Floor for Small Biotech Drugs.
bt. Specified Manufacturer
We propose to define “Specified Manufacturer” as having the meaning set forth in section 1860D-14C(g)(4)(B)(ii) of the Act, as determined by CMS for the purposes of the Manufacturer Discount Program in accordance with §§ 423.2716, 423.2720, and 423.2724.
bu. Starting Point
We propose to define “starting point” as the numerical dollar amount used by CMS in developing an initial offer in accordance with proposed § 429.510(d) that is then adjusted by CMS based on section 1194(e)(2) factors to determine the preliminary price, per the process described in proposed § 429.510(e).
bv. Temporary Floor for Small Biotech Drugs
We propose to define “Temporary Floor for Small Biotech Drugs” as having the meaning set forth in § 429.440(b)(3). We refer readers to proposed § 429.440(b)(1) and (2) (and related discussion in section II.E.9.b. of this proposed rule) for CMS' proposals for the process for a Primary Manufacturer to request consideration and CMS' determination of eligibility for the Temporary Floor for Small Biotech Drugs.
bw. Therapeutic Advance
We propose to define “therapeutic advance” as a demonstrated improvement in one or more outcomes or other clinical considerations for each identified condition of a selected drug as compared to its therapeutic alternative(s). For purposes of the Negotiation Program, anytime CMS considers therapeutic advance, CMS would consider the extent to which the drug represents a therapeutic advance at the time of consideration based on all available information at such time of consideration
bx. Therapeutic Alternative
We propose to define “therapeutic alternative” as a pharmaceutical product or group of pharmaceutical products other than the selected drug that may be used to treat the same condition or disease state as the selected drug.
by. Total Allowed Charges
We propose to define “total allowed charges” as the amount that is inclusive of the beneficiary coinsurance and Medicare payment for the covered Part B item or service paid for under part B of Title XVIII of the Act, without a sequestration payment adjustment applied.
bz. Total Expenditures
We propose to define “total expenditures” as having the meaning set forth in section 1191(c)(5) of the Act. We refer readers to § 429.120 and section II.B.5. of this proposed rule for CMS' proposals for calculating total expenditures under Part D and total expenditures under Part B that meet this statutory definition.
ca. Total Expenditures Measurement Period
Sections 1192(d)(1)(A) and (d)(1)(B) of the Act require that CMS calculate total expenditures under Part D and Part B, respectively, using data from the most recent 12-month period for which data are available prior to the selected drug publication date with respect to an initial price applicability year, but ending no later than October 31 of the year prior to the year of such drug publication date. To describe this 12-month period, we propose to define “total expenditures measurement period” as the 12-month period ending on October 31 of the year prior to the year of the selected drug publication date with respect to an initial price applicability year.
cb. Total Gross Covered Prescription Drug Costs
Section 1191(c)(5) of the Act specifies that the term “total gross covered prescription drugs costs” is defined at section 1860D-15(b)(3) of the Act. The term “total gross covered prescription drug costs” does not appear at section 1860D-15(b)(3) of the Act, but section 1860D-15(b)(3) of the Act does define the term “gross covered prescription drug costs,” and § 423.308 codifies this term. We therefore propose to define “total gross covered prescription drug costs” as having the meaning given the term “gross covered prescription drug costs” set forth at 42 CFR 423.308.
cc. Unit
We propose to define “unit” as having the meaning set forth in section 1191(c)(6) of the Act.
cd. Unmet Medical Need
We propose to define “unmet medical need” as a circumstance in which the relevant disease or condition is one for which no other treatment options exist, or existing treatments do not adequately address the disease or condition. For purposes of the Negotiation Program, anytime CMS considers an unmet medical need, CMS would consider the extent to which the drug addresses an unmet medical need at the time of consideration based on all available information at such time of consideration.
ce. Wholesale Acquisition Cost (WAC) Unit Price
We propose to define “Wholesale Acquisition Cost (WAC) unit price” as the manufacturer's list price for the drug or biological product to wholesalers or
( printed page 36243)
direct purchasers in the United States, not including prompt pay or other discounts, rebates or reductions in price, for the most recent month for which the information is available, as reported in wholesale price guides or other publications of drug or biological product pricing data (as defined in section 1847A(c)(6)(B) of the Act). The WAC unit price is reported at the NDC-11 level.
3. Limitation on Review (§ 429.30)
Section 1198 of the Act establishes that there shall be no administrative or judicial review of any of the following: (1) the determination of a unit, with respect to a drug or biological product, pursuant to section 1191(c)(6) of the Act; (2) the selection of drugs under section 1192(b) of the Act, the determination of negotiation-eligible drugs under section 1192(d) of the Act, the determination of qualifying single source drugs under section 1192(e) of the Act, and the application of the Biosimilar Delay under section 1192(f) of the Act; (3) the determination of a MFP under subsection (b) or (f) of section 1194 of the Act; and (4) the determination of renegotiation-eligible drugs under section 1194(f)(2) of the Act and the selection of renegotiation-eligible drugs under section 1194(f)(3) of the Act. CMS proposes to codify these limitations on review in proposed § 429.30.
B. Identification of Selected Drugs (§§ 429.100 Through 429.135)
Section 1192 of the Act establishes the requirements governing the publication of the list of selected drugs for an initial price applicability year, the identification of selected drugs, ranking of negotiation-eligible drugs, and the identification of qualifying single source drugs. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example with respect to initial price applicability year 2028, section 30 of the Negotiation Program Guidance. With respect to initial price applicability years beginning with initial price applicability year 2029, we are proposing to codify these steps in an order reflecting the sequence of the statutory provisions which these sections are implementing, with proposed revisions as noted in this section, in proposed §§ 429.100 through 429.135.
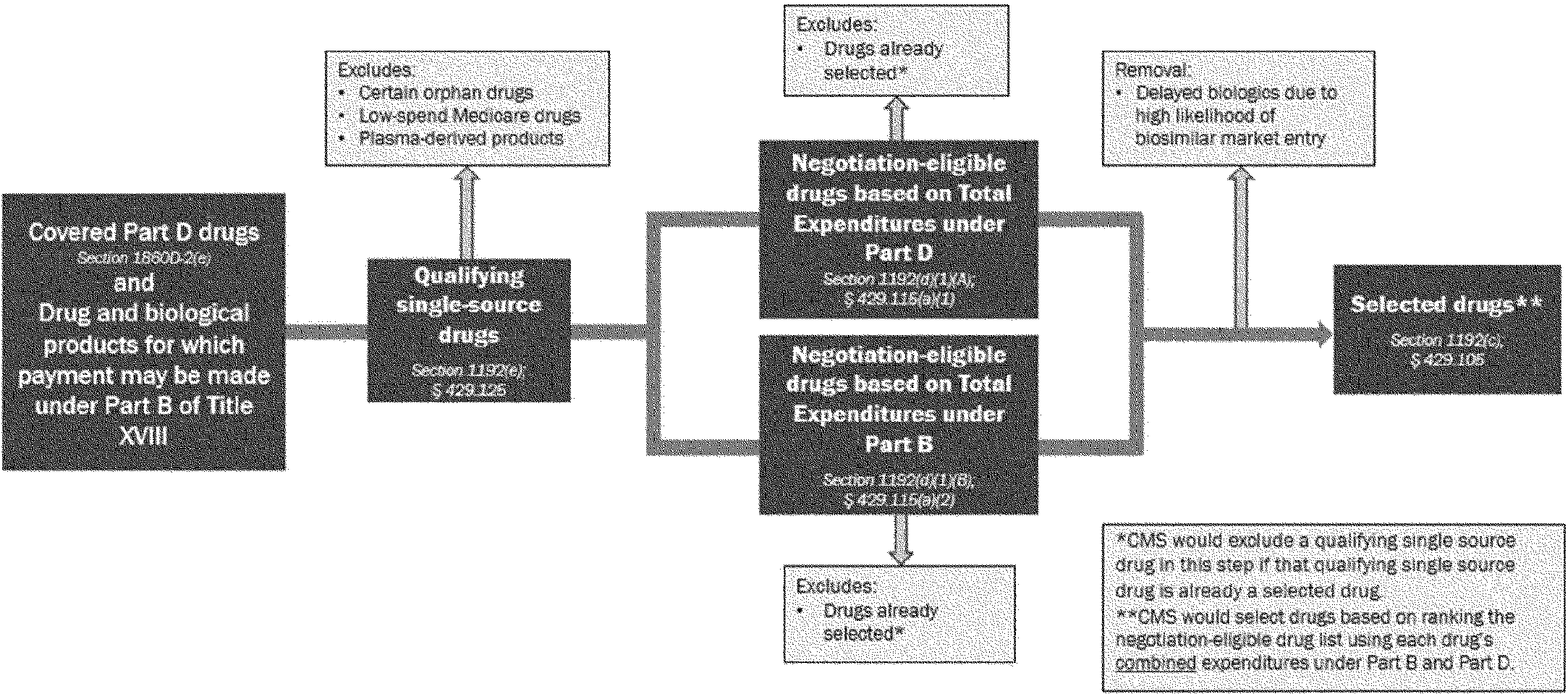
Beginning with respect to initial price applicability year 2029 and in accordance with section 1192 of the Act, we propose in §§ 429.100 through 429.135 to codify the policies for identification of selected drugs described in sections 30 and 40.2 of the Negotiation Program Guidance, subject to proposed modifications as noted herein. As a matter of program operations, we would first identify qualifying single source drugs with respect to each initial price applicability year. As a part of this identification process, CMS would exclude certain drugs as proposed in § 429.125(e). Next, we would identify negotiation-eligible drugs using total expenditures under Part B or Part D of Title XVIII of the Act, as applicable and calculated as set forth in proposed § 429.120, to identify qualifying single source drugs that are Part B high spend drugs, Part D high spend drugs, or both, as proposed in § 429.115. (In these steps, we would also exclude drugs that are already selected drugs in accordance with section 1192(d)(3) of the Act.) As proposed in § 429.105(a), we would rank these negotiation-eligible drugs for an initial price applicability year according to the total expenditures for such drugs. In accordance with section 1192(a) of the Act and subject to the section 1192(f) of the Act (which permits the delay in the selection and negotiation of biological products for biosimilar market entry when certain requirements are met consistent with proposed § 429.110, hereinafter “Biosimilar Delay”), we propose at § 429.105(c) to select up to 20 negotiation-eligible drugs with the highest total expenditures under Part B and Part D of Title XVIII of the Act for negotiation for initial price applicability year 2029 and each initial price applicability year thereafter, and publish the list of selected drugs as proposed at § 429.100. We may also select a drug or drugs for renegotiation based on criteria discussed in detail in section II.G.3. of this proposed rule and in proposed § 429.610.
Finally, as proposed in § 429.100, we would publish the list of drugs selected for negotiation, including the list of drugs selected for renegotiation, if any, not later than the selected drug publication date. We are also proposing to publish a list of the up to 30 top negotiation-eligible drugs (including the up to 20 selected drugs) ranked by combined total expenditures under Part B and Part D. Detailed descriptions of these proposals for initial price applicability year 2029 and each initial price applicability year thereafter is included later in this section. Figure 1 provides a visual depiction of this proposed process.
Figure 1—Diagram of Proposed Process for Selecting Drugs for Negotiation for Initial Price Applicability Years Beginning With Initial Price Applicability Year 2029
( printed page 36244)
1. Publication of the Selected Drug List (§ 429.100)
Section 1192(a)(4) of the Act requires that, not later than the selected drug publication date with respect to the initial price applicability year, in accordance with section 1192(b) of the Act, the Secretary shall select and publish a list of, with respect to the initial price applicability year 2029 or a subsequent year, 20 negotiation-eligible drugs, as described in section 1192(d)(1) of the Act, with respect to such year (or, all (if such number is less than 20) such negotiation-eligible drugs with respect to such year). Proposed § 429.20 defines the term “selected drug publication date” to have the meaning set forth in section 1191(b)(3) of the Act, which provides that the term “selected drug publication date” means, with respect to each initial price applicability year, February 1 of the year that begins 2 years prior to such year. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 30.4 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
We are proposing at § 429.100(a) to codify the requirement at section 1192(c)(1) of the Act that each drug included on the selected drug list [12]
for an initial price applicability year is a selected drug with respect to such initial price applicability year and each subsequent year unless and until CMS makes a determination in accordance with proposed § 429.135(a) that such drug will be deselected (as described in further detail in section II.B.6.d. of this proposed rule). We are proposing at § 429.100(b) to codify the requirement that CMS publish the selected drug list and the drugs selected for renegotiation, if any, for each initial price applicability year beginning with initial price applicability year 2029, no later than the selected drug publication date with respect to the initial price applicability year. For example, for initial price applicability year 2029, we would publish this information no later than February 1, 2027. As proposed in § 429.100(b)(1), the selected drug list would include the 20 (or all, if such number is less than 20) drugs payable under Part B, covered under Part D, or both, selected for negotiation for the initial price applicability year as determined in § 429.105(c) and discussed in section II.B.2 of this proposed rule. As proposed in § 429.100(b)(2), we would also publish the list of drugs selected for renegotiation, if any, as set forth in proposed § 429.610.
For each selected drug, we are proposing at § 429.100(b)(3)(i) to add to the MFP file no later than the selected drug publication date the active moiety, active ingredient, antigen component, or, in the case of a potential qualifying single source drug identified under the general fixed combination drug policy proposed at § 429.125(b)(4), the distinct combination of active moieties, active ingredients, or antigen components,[13]
as applicable, identified as set forth in proposed § 429.125(b). For a potential qualifying single source drug identified under § 429.125(b)(4)(i), we are proposing to publish the shared active moiety/active ingredient identified under § 429.125(b)(4)(i), plus any additional active moiety/active ingredient included in new formulations of such potential qualifying single source drug. We are proposing at § 429.100(b)(4)(i) to take the same approach for each drug selected for renegotiation, if any, except we would publish the active moiety/active ingredient previously identified for the initial price applicability year for which the drug was originally selected for negotiation. We are proposing at § 429.100(b)(3)(ii) and (b)(4)(ii) to add to the MFP file no later than the selected drug publication date the NDC-11s identified in accordance with § 429.100(c)(1) and the corresponding NDC-9s and HCPCS codes, as applicable, for the selected drug and the drug selected for renegotiation, if any. For drugs selected for renegotiation, the NDC-11s (and corresponding NDC-9s
( printed page 36245)
and HCPCS codes) added to the MFP file would also reflect information previously submitted by the Primary Manufacturer, including submissions in accordance with proposed § 429.100. At § 429.100(c), we propose the process we would use to identify the list of NDC-11s described in the prior sentences for each selected drug and each drug selected for renegotiation, if any. As proposed at § 429.100(f), the agency's list of NDC-11s would be used in the administration of the Negotiation Program, including to identify the NDC-11s of the selected drug that are subject to the negotiation process set forth in proposed subpart F and the renegotiation process set forth in proposed subpart G (as applicable), identify the NDC-11s of the selected drug to which the MFP (if one is agreed to by CMS and the Primary Manufacturer) applies for the price applicability period, to calculate the ceiling set forth in proposed § 429.410 for drugs selected for negotiation, to calculate the ceiling set forth in proposed § 429.620(b) for drugs selected for renegotiation, and to calculate how to apply the MFP, if one is agreed to by CMS and the Primary Manufacturer, and to the extent data are available to support such calculations, across dosage forms and strengths set forth in proposed § 429.700 for selected drugs and proposed § 429.600(b)(2) for drugs selected for renegotiation.
To identify the list of NDC-11s of the selected drug, including for a drug selected for renegotiation, set forth at proposed § 429.100(c), we propose at § 429.100(c)(1) to first identify NDC-11s associated with the NDA(s)/BLA(s) of the selected drug. We would compile all NDC-11s belonging to the selected drug associated with HCPCS codes that appear on NDC-HCPCS code crosswalks published by CMS [14]
for the most recent quarter in the total expenditures measurement period (as such term is defined in proposed § 429.20), and all NDC-11s belonging to the selected drug that had Part D PDE utilization in the total expenditures measurement period. We would also identify any additional NDC-11s associated with the NDA(s)/BLA(s) of the selected drug as found in recent updates of the NDC Structured Product Labeling (SPL) Data Elements file (NSDE) file or the NDC Directory (including its NDC Excluded Drugs Database file). In section 30.4 of the Negotiation Program Guidance, we stated that we will remove any NDC-11s for which CMS has evidence suggesting a lack of coverage under Part D and Part B. Based on lessons learned from policy implementation in initial price applicability years 2026 through 2028, we are proposing to remove such requirement for initial price applicability year 2029 and subsequent years. Starting with a more comprehensive list of NDC-11s holds utility for CMS and Primary Manufacturers, as it reduces the number of NDC-11s that a Primary Manufacturer must identify as missing from the list, as required in proposed § 429.100(d)(1). We would publish the selected drug list, as well as the list of drugs selected for renegotiation, in a form and manner of CMS' choosing, which may be on the CMS website.
We are proposing at § 429.100(c)(2) to transmit the list of NDC-11s identified at proposed § 429.100(c)(1) to the Primary Manufacturer. As proposed at § 429.100(c)(3), we may revise our list of NDC-11s of each selected drug, including without limitation using information submitted by the Primary Manufacturer in accordance with proposed § 429.100.
In accordance with a Primary Manufacturer's responsibility under section 1193(a)(4)(B) of the Act and under the Negotiation Program Agreement (set forth in proposed § 429.200 and described in section II.C.1. of this proposed rule), we propose in § 429.100(d) that a Primary Manufacturer must review the list of NDC-11s provided by CMS at proposed § 429.100(c) and provide information on each NDC-11 on the list of NDC-11s that make up a selected drug as a part of their data submission. More specifically, we propose at § 429.100(d) that a Primary Manufacturer must review the list of NDC-11s and provide proposed revisions to the list, as needed, by adding any NDC-11s associated with the NDA(s)/BLA(s) of the selected drug that do not appear on the agency's list of NDC-11s of the selected drug, including any missing NDC-11s of a Secondary Manufacturer. A Primary Manufacturer must also provide identifying information for any NDC-11 that appears on the list of NDC-11s, including any NDC-11s added by the Primary Manufacturer, on whether NDC-11(s): are for products distributed by or under the name of a private label distributor; are not manufactured, marketed, controlled or sold by the Primary Manufacturer or a Secondary Manufacturer; represent a sample package; represent an inner package or an outer package; and whether an NDC-11 has been discontinued. As described in proposed § 429.100(c)(3), we may revise the list of NDC-11s that make up the selected drug based on this information submitted by the Primary Manufacturer.
In accordance with a Primary Manufacturer's responsibility under section 1193(a)(5) of the Act and under the Negotiation Program Agreement (set forth in proposed § 429.200), we propose in § 429.100(e) that a Primary Manufacturer has an ongoing obligation to report, at least 30 calendar days prior to the change taking effect, any changes to the information provided in § 429.100(d) to ensure the list of NDC-11s of the selected drug identified in accordance with proposed § 429.100(c) remains complete and accurate. For example, under proposed § 429.100(e), a Primary Manufacturer must report to CMS any new NDC-11s of the selected drug at least 30 days prior to their first marketed date by or on behalf of the Primary Manufacturer or any Secondary Manufacturer(s) of such selected drug. Failure to provide timely reporting of changes to the list of NDC-11s of the selected drug as described in proposed § 429.100(e) may be considered a violation of the Negotiation Program Agreement under section 1193(a)(5) of the Act and proposed § 429.200(b).
Since the Negotiation Program's inception, interested parties have recommended greater transparency into the process for selecting drugs. In response to these recommendations and in accordance with policy established in the Negotiation Program Guidance, we published a list of the 50 top negotiation-eligible drugs for initial price applicability year 2028 (including the 15 selected drugs for initial price applicability year 2028).[15]
To harmonize the request from interested parties for greater transparency into the process for selecting drugs with CMS operations, for initial price applicability year 2029 and subsequent years, we are proposing to publish a list of the up to 30 top negotiation-eligible drugs (including the up to 20 selected drugs) ranked by combined total expenditures under Part B and Part D, as determined under proposed § 429.105(a), and information on the NDC-9s, NDC-11s, and HCPCS codes for these negotiation-eligible drugs, as applicable and to the extent feasible. The purpose of publishing a list of negotiation-eligible drugs beyond selected drugs was, and remains, to promote transparency in the drug selection process. The conditions that determine which drugs meet the statutory requirements for a drug to become a qualifying single source drug, negotiation-eligible drug, or selected
( printed page 36246)
drug for a given initial price applicability are not static. Such list was not, and is not, intended to predict or replicate the selected drug list for future initial price applicability years. The honed focus on the up to 30 top drugs would continue to provide transparency into the drug selection process. We believe the prior policy of publishing negotiation-eligible drugs with rankings lower than 30 (that is, #31 through #50) provided less meaningful transparency into the drug selection process for a given initial price applicability year, as identifying such drugs provides little insight into the criteria and conditions that were material to the identification of the selected drug list for that initial price applicability year. Finally, consistent with prior policy, we propose that the list of top drugs based on combined total expenditures would reflect the removal of negotiation-eligible drugs that qualify for the Biosimilar Delay.
2. Selection of Drugs for Negotiation (§ 429.105)
Section 1192(b)(1)(A) of the Act requires that, in carrying out section 1192(a) of the Act, the Secretary shall, with respect to an initial price applicability year, rank negotiation-eligible drugs, as described in section 1192(d)(1) of the Act, according to the total expenditures for such drugs under parts B and D of Title XVIII, as determined by the Secretary, during the most recent period of 12 months prior to the selected drug publication date (but ending not later than October 31 of the year prior to the year of such drug publication date), with respect to such year, for which data are available, with the negotiation-eligible drugs with the highest total expenditures being ranked the highest. Section 1192(b)(1)(B) of the Act requires that the Secretary select from such ranked drugs with respect to such initial price applicability year the negotiation-eligible drugs with the highest such rankings. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 30.3 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
We are proposing at § 429.105 to select 20 (or all, if such number is less than 20) negotiation-eligible drugs for negotiation for each initial price applicability year.
First, with respect to an initial price applicability year, we are proposing at § 429.105(a) to rank the list of negotiation-eligible drugs identified at proposed § 429.115 by combined total expenditures under both Part B and Part D in descending order: the negotiation-eligible drug with the highest total expenditures under Part B and Part D would be listed first, and the negotiation-eligible drug with the lowest total expenditures under Part B and Part D would be listed last (the proposed methodology for the calculation of total expenditures under Part B and total expenditures under Part D is described in proposed § 429.120 and section II.B.5. of this proposed rule). If a negotiation-eligible drug appears on both the Part D high-spend drug list and Part B high-spend drug list (set forth in proposed § 429.115(a)(1) and (a)(2), respectively, and described in section II.B.4. of this proposed rule), it would receive only one ranking for purposes of selection, according to its combined total expenditures under both Part B and Part D. If a negotiation-eligible drug appears on only one high-spend list, CMS would still combine total expenditures under both Part B and Part D.
Second, with respect to an initial price applicability year, we are proposing at § 429.105(b) to remove any biological products that qualify for delayed selection under section 1192(f) of the Act, as proposed at § 429.110 and described in section II.B.3. of this proposed rule.
Finally, we propose at § 429.105(c) to select for negotiation the 20 (or all, if such number is less than 20) highest ranked negotiation-eligible drugs remaining on the ranked list for the initial price applicability year. In guidance for initial price applicability years 2026, 2027 and 2028, including, for example, section 30.3 of the Negotiation Program Guidance, we established that for initial price applicability years 2026, 2027, and 2028, in the event that two or more negotiation-eligible drugs had the same total expenditures to the dollar, and such total expenditures were the 10th or 15th highest among negotiation-eligible drugs, as applicable for the initial price applicability year, we will rank those negotiation-eligible drugs based on which drug had the earlier approval or licensure date, as applicable, associated with the earliest-approved FDA application belonging to the NDA/BLA holder and containing the drug's active moiety/active ingredient, and select based on that ranking until there were 10 or 15 (as applicable) selected drugs (or until all drugs were selected, if the number of negotiation-eligible drugs was less than 10 or 15, as applicable). In this proposed rule, we are proposing to modify this methodology. We propose that to determine whether two or more negotiation-eligible drugs have the same total expenditures, and such total expenditures are the 20th highest among negotiation-eligible drugs (or the highest, if the number is less than 20), we would evaluate such total expenditures to the cent, rather than to the dollar as under prior policy. We believe that determining total expenditures to the cent, rather than the dollar, is more precise for purposes of determining the selected drug list. For such drugs with the same combined total expenditures under Part B and Part D to the cent, we would continue to rank those negotiation-eligible drugs based on which drug has the earliest-approved FDA application belonging to the NDA/BLA holder and containing the drug's active moiety/active ingredient, and select based on that ranking until there are 20 selected drugs (or until all drugs are selected, if the number of negotiation-eligible drugs is less than 20).
3. Request for a Biosimilar Delay (§ 429.110)
a. Overview of the Requirements for a Delay in the Selection and Negotiation of Certain Biological Products With High Likelihood of Biosimilar Market Entry
Section 1192(b)(1)(C) of the Act requires the Secretary to remove from the ranked list of negotiation-eligible drugs (described in proposed § 429.105 and section II.B.2. of this proposed rule) any negotiation-eligible drug for which the inclusion on the selected drug list is delayed in accordance with section 1192(f) of the Act. Specifically, section 1192(f)(1)(B) of the Act allows the manufacturer of a biosimilar biological product (defined at proposed § 429.20 as the “Biosimilar Manufacturer” of a Biosimilar) to submit a request, prior to the selected drug publication date for an initial price applicability year, for CMS' consideration to delay the inclusion of a negotiation-eligible drug that includes the reference product for the Biosimilar (defined at proposed § 429.20 as a “Reference Drug”) on the selected drug list for such given initial price applicability year (which we refer to as a “Biosimilar Delay”).
Section 1192(f) of the Act provides for two potential requests for a Biosimilar Delay: (1) a request to delay the inclusion of a Reference Drug by one initial price applicability year (“Initial Delay Request” as defined in proposed § 429.20) under section 1192(f)(1)(B)(i)(I) of the Act; and (2) a request to delay the inclusion of a Reference Drug for which an Initial Delay Request has been granted for a second initial price applicability year
( printed page 36247)
(“Additional Delay Request”) under section 1192(f)(1)(B)(i)(II) of the Act. Together, CMS refers to an Initial Delay Request and an Additional Delay Request as “Biosimilar Delay Requests” as defined in proposed § 429.20. Proposed § 429.110(b) through (f) address the requirements for a Biosimilar Manufacturer to submit a Biosimilar Delay Request and for CMS to determine if the inclusion of the Reference Drug on the selected drug list should be delayed due to such Biosimilar Delay Request. As set forth in proposed § 429.110(a), for purposes of the provisions at proposed § 429.110 and in our discussion of this section herein, all references to “marketed” or “marketing” mean Bona Fide Marketing as defined in proposed § 429.20 and set forth at proposed § 429.130(a). We discuss Bona Fide Marketing further in section II.B.6.d. of this proposed rule.
Biosimilar Manufacturers that believe that the Reference Drug of their Biosimilar may be a selected drug for an initial price applicability year may submit an Initial Delay Request for the first year and an Additional Delay request for the second year, and CMS would disregard that request if the Reference Drug would not, in fact, be a selected drug for an initial price applicability year. Biosimilar Manufacturers are encouraged to consult publicly available data on expenditures for drugs payable under Part B and/or covered under Part D, including data published by CMS, including but not limited to data on the Medicare Part B Drug Spending Dashboard [16]
and the Medicare Part D Drug Spending Dashboard,[17]
which may allow them to determine the likelihood that a given drug may be a selected drug.
As discussed in further detail in section IV. of this proposed rule, we are also proposing revisions to a currently approved information collection for a manufacturer to submit an Initial Delay Request, titled the Negotiation Program Drug Selection for Initial Price Applicability Year 20XX under Section 11001 and 11002 of the Inflation Reduction Act Information Collection Request (ICR) (CMS-10844, OMB 0938-1443) (hereinafter, the “Drug Selection ICR”), for a 60-day public comment period concurrently with this proposed rule. A form and manner for submitting a Biosimilar Delay Request, consistent with proposed § 429.110(f), would be specified in the ICR for an initial price applicability year for an Initial Delay Request or an Additional Delay Request. As discussed in further detail in the accompanying 60-day package, we are including questions specific to an Initial Delay Request only within the ICR because CMS did not grant an Initial Delay Request for initial price applicability year 2028 and thus we are not including questions pertaining to submitting an Additional Delay Request for initial price applicability year 2029. We will expand the collection to include questions pertaining to an Additional Delay Request when necessary for an upcoming initial price applicability year when there is a Biosimilar Manufacturer that would be eligible to submit an Additional Delay Request after the granting of an Initial Delay Request. Information submitted in a Biosimilar Delay Request that is trade secret or confidential commercial or financial information will be protected from disclosure if the information meets the requirements set forth under Exemptions 3 and/or 4 of the Freedom of Information Act (FOIA) (5 U.S.C. 552(b)(3), (4)).
For an Initial Delay Request, if we determine that an otherwise negotiation-eligible drug should be delayed from selection because of the requirements proposed in § 429.110(c), but the Biosimilar is not licensed and marketed based on the requirements proposed in § 429.110(h)during the Initial Delay Period (which we propose to define in § 429.20 as the time period between (1) the selected drug publication date for the initial price applicability year for which the Reference Drug otherwise would have been included on the selected drug list but for the successful Initial Delay Request, and (2) the selected drug publication date with respect to the initial price applicability year that is 1 year after the initial price applicability year for which the Reference Drug otherwise would have been included on the selected drug list but for the successful Initial Delay Request), the Biosimilar Manufacturer would have the opportunity to submit an Additional Delay Request consistent with proposed § 429.110(e). If the Biosimilar Manufacturer fails to submit an Additional Delay Request or submits an Additional Delay Request that we determine does not meet all the requirements proposed in § 429.110(e), as proposed in § 429.110(h)(1)(ii), the Reference Drug would be included on the selected drug list for the initial price applicability year that is 1 year after the initial price applicability year for which the Reference Drug would have been included on the selected drug list if not for the successful Initial Delay Request. However, we would not include the Reference Drug on such list if another biosimilar of the Reference Drug is marketed before the publication date of the list.
If the Biosimilar named in a successful Additional Delay Request is not licensed and marketed during the Second Delay Period (which we propose to define in § 429.20 as the time period between (1) the publication date of the selected drug list for initial price applicability year that is 1 year after the initial price applicability year for which the Reference Drug would have been included on the selected drug list but for the successful Initial Delay Request, and (2) the publication date of the selected drug list for initial price applicability year that is 2 years after the initial price applicability year for which the Reference Drug would have been included on the selected drug list but for the successful Initial Delay Request), as proposed in § 429.110(h)(2), the Reference Drug would be included on the selected drug list for the initial price applicability year that is 2 years after the initial price applicability year for which the Reference Drug would have been included on the selected drug list if not for the successful Initial Delay Request(s). However, if another biosimilar of the Reference Drug is marketed prior to the publication date of such list, we would not include the Reference Drug on the list.
Additionally, as proposed in § 429.110(i)(1), if CMS delayed the selection and negotiation of a Reference Drug for 1 or 2 years, but the Biosimilar was not licensed and marketed, and the Reference Manufacturer agrees to an MFP for the Reference Drug, the Reference Manufacturer would owe a rebate to the Federal Supplementary Medical Insurance Trust Fund for drugs payable under Part B or the Medicare Prescription Drug Account for drugs covered under Part D for the years that the manufacturer would have provided access to the MFP for the Reference Drug but for the successful Biosimilar Delay Requests. Consistent with section 1192(f)(4) of the Act and as described in section II.B.3.c. of this proposed rule, proposed § 429.110(i) includes the proposed requirements for the calculation of the rebate.
Consistent with section 1198(2) of the Act and proposed § 429.30, there would no administrative or judicial review of CMS' determinations under section 1192(f) of the Act and in proposed § 429.110 regarding a Biosimilar Delay Request.
( printed page 36248)
b. Requirements for Granting a Biosimilar Delay Request (§ 429.110(c) Through (f))
Section 1192(f)(1)(B)(ii)(I) of the Act requires that the request for the delay be made by the Biosimilar Manufacturer and cannot be initiated by a separate party, such as CMS or the Reference Manufacturer. The Biosimilar Manufacturer, as defined in proposed § 429.20, that is specifically eligible to submit the request is the BLA holder for the Biosimilar or, if the Biosimilar has not yet been licensed, the sponsor of the BLA submitted for review by the FDA. Also included in the definition of “Biosimilar Manufacturer” at proposed § 429.20, if neither the Biosimilar has been licensed nor the BLA has been submitted to FDA, the Biosimilar Manufacturer eligible to submit the request is the organization planning to be the sponsor of the BLA submitted for review by FDA. This approach, which is consistent with the policies for implementation as described in sections 30.3.1 through 30.3.1.5 of Negotiation Program Guidance, is appropriate because: (1) it clearly identifies one manufacturer that may submit a Biosimilar Delay Request for a given Biosimilar, avoiding the possibility that CMS would receive two such requests naming the same Biosimilar for the same initial price applicability year; and (2) the status of the application for licensure for the Biosimilar is material to CMS' consideration of a Biosimilar Delay Request, as described in proposed § 429.110. For both an Initial Delay Request and an Additional Delay Request, certain requirements must be met for CMS to grant such requests. These requirements are included in proposed § 429.110(c) for an Initial Delay Request and proposed § 429.110(e) for an Additional Delay Request.
Section 1192(f)(1)(B)(ii)(I) and (II) of the Act requires the Biosimilar Manufacturer to make the request prior to the selected drug publication date for the initial price applicability year for which the Biosimilar Manufacturer is requesting a Biosimilar Delay. As such, we are proposing at § 429.110(f) that a Biosimilar Manufacturer may submit to CMS a request for a Biosimilar Delay at the time and in a form and manner specified by CMS. Consistent with the process and timeline for previous initial price applicability years, CMS intends to collect requests via the CMS Health Plan Management System (CMS HPMS) and provide for a 30-day submission period as discussed in the Drug Selection ICR. We will not consider late or incomplete submissions. Upon receipt of a complete Biosimilar Delay Request, CMS will consider whether the requirements are met, as applicable, in proposed 429.110(c) for an Initial Delay Request or proposed § 429.110(e) for an Additional Delay Request.
With respect to Initial Delay Requests, we would first determine if the proposed requirements under proposed § 429.110(c)(1) have been met. Section 1192(f)(1)(A) of the Act and, as described in proposed § 429.110(c)(1)(i), requires that the Reference Drug would be an extended-monopoly drug, as defined in section 1194(c)(4) of the Act and proposed § 420.20, included on the selected drug list for the initial price applicability year, absent the Biosimilar Delay. For Initial Delay Requests, this means that the Reference Drug must have received its initial BLA licensure at least 12 years, but fewer than 16 years, prior to the start of the relevant initial price applicability year. Section 1194(c)(4)(B)(ii) of the Act specifies that selected drugs for which a manufacturer had an agreement under the Negotiation Program for an initial price applicability year prior to 2030 are excluded from the definition of extended-monopoly drugs (definition proposed at § 429.20). Importantly, however, an Initial Delay Request must be submitted by a Biosimilar Manufacturer before the selected drug publication date for an initial price applicability year and before the Reference Manufacturer would have entered into an agreement under the Negotiation Program. Therefore, we continue to believe the exception to the definition of “extended-monopoly drug” in section 1194(c)(4)(B)(ii) of the Act would not apply at the time that a delay would be requested for initial price applicability year 2029. Accordingly, we believe the Biosimilar Delay Request process under section 1192(f) of the Act is applicable for future initial price applicability years. As such, Biosimilar Manufacturers may submit an Initial Delay Request for initial price applicability year 2029, provided that the Reference Drug named in the request would have been licensed for at least 12 years but fewer than 16 years prior to the start of the initial price applicability year beginning on January 1, 2029.
Additionally, to qualify for an Initial Delay Request, section 1192(f) of the Act requires the following (as proposed in § 429.110(c)):
In accordance with section 1192(f)(1)(A) of the Act and as described in proposed § 429.110(c)(1)(ii), the Reference Drug must include the reference product identified in the Biosimilar's application for licensure under section 351(k) of the PHS Act that has been approved or accepted for review by FDA. We note that to grant a Biosimilar Delay Request, the licensure application for the Biosimilar does not need to include all of the dosage forms, strengths, and indications for which the Reference Drug has received approval. With respect to the reference product, the Initial Delay Request may list the brand name and/or the name of the reference product's active ingredient.
In accordance with section 1192(f)(2)(D)(iii) of the Act and as described in proposed § 429.110(c)(iii), a Biosimilar Delay Request would not be granted if more than 1 year has elapsed since the licensure of the Biosimilar and marketing of the Biosimilar has not commenced.
In accordance with section 1192(f)(2)(D)(iv) of the Act and as described in proposed § 429.110(c)(1)(iv)(A), the Biosimilar Manufacturer must not be the same as the Reference Manufacturer and must not be treated as being the same under section 1192(f)(1)(C) of the Act. For the purposes of this determination, all persons treated as a single employer under subsection (a) or (b) of section 52 of the Internal Revenue Code (IRC), or in a partnership, shall be treated as one manufacturer in accordance with section 1192(f)(1)(C) of the Act. For the purposes of this determination, “partnership” (as proposed at § 429.20) is defined at section 1192(f)(1)(C)(ii) of the Act as a syndicate, group, pool, joint venture, or other organization through or by means of which any business, financial operation, or venture is carried on by the Reference Manufacturer and the Biosimilar Manufacturer.
In accordance with section 1192(f)(2)(D)(iv) of the Act and as described in proposed § 429.110(c)(1)(iv)(B), the Biosimilar Manufacturer and the Reference Manufacturer must not have entered into an agreement that—
++ Requires or incentivizes the Biosimilar Manufacturer to submit a Biosimilar Delay Request; or
++ Directly or indirectly restricts the quantity of the Biosimilar that may be sold in the United States over a specified period of time.
We would consider any agreement between the Biosimilar Manufacturer and the Reference Manufacturer that directly or indirectly restricts the quantity of the Biosimilar that the Biosimilar Manufacturer may sell during any period of time on or after the selected drug publication date for the initial price applicability year for which the Biosimilar Manufacturer is requesting a Biosimilar Delay, as failing to meet this requirement.
( printed page 36249)
Once we determine the requirements proposed in § 429.110(c)(1) are met, we would then determine if there is a high likelihood, as required in section 1194(f)(1)(A) of the Act and as proposed in § 429.110(c)(2), that the Biosimilar will be licensed and marketed before the date that is 2 years after the statutorily defined selected drug publication date for the initial price applicability year for which the Reference Drug would be included on the selected drug list absent a successful Initial Delay Request (“High Likelihood Deadline,” as defined in proposed § 429.20). For example, the High Likelihood Deadline for an Initial Delay Request for initial price applicability year 2029 would be February 1, 2029. Specifically, in accordance with section 1192(f)(3) of the Act and consistent with implementation of the policies in section 30.3.1.3 of Negotiation Program Guidance Program, we propose in § 429.110(d) that there is a high likelihood the Biosimilar will be licensed and marketed before the High Likelihood Deadline if each of the following criteria are met:
An application for licensure under section 351(k) of the PHS Act for the Biosimilar has been accepted for review or licensed by FDA.
Clear and convincing evidence that the Biosimilar will be marketed before the High Likelihood Deadline.
We propose at § 429.110(d)(1) that CMS will specify the due date by which the application for licensure must be accepted for review or approved by the FDA, which will be a date before the selected drug publication date for the initial price applicability year for which the Biosimilar Manufacturer requests a Biosimilar Delay in order to permit sufficient time for CMS to review the information and finalize the selected drug list prior to publishing the selected drug list for the initial price applicability year. This would enable CMS to use the most recent possible data to make this determination, while still allowing for sufficient time for such requests to inform the selected drug list prior to the selected drug publication date as required by section 1192(a) of the Act. If the Biosimilar's application for licensure has not been accepted for review by the specified date, including in the case where the Biosimilar Manufacturer submitted an application for licensure that has not been accepted for review by FDA or for which a filing determination is pending, we would deny the Initial Delay Request. Additionally, CMS would consider an application for licensure under section 351(k) of the PHS Act that has been accepted for review and received a complete response letter from the FDA to meet the section 1192(f)(3)(A) requirement that an application for licensure under section 351(k) for the biosimilar biological product has been accepted for review by FDA.
To demonstrate clear and convincing evidence that the Biosimilar will be marketed before the High Likelihood Deadline, we propose at § 429.110(d)(2) that the Biosimilar Delay Request must include information to demonstrate both: (1) that patents related to the Reference Drug are unlikely to prevent the Biosimilar from being marketed; and (2) that the Biosimilar Manufacturer will be operationally ready to market the Biosimilar. These requirements address the two primary contributing factors to delays in marketing of biosimilars approved in the U.S. to date, and so we believe that evidence showing that a Biosimilar meets these two requirements is sufficient to establish clear and convincing evidence that the Biosimilar will be marketed.
First, regarding the proposal at § 429.110(d)(2)(i) that the Biosimilar Delay Request must clearly demonstrate that patents related to the Reference Drug are unlikely to prevent the Biosimilar from being marketed before the High Likelihood Deadline: we will only consider patents relating to the reference product included in the Reference Drug that are applicable to the Biosimilar. For example, if a Biosimilar Manufacturer has obtained licensure with biosimilar labeling that omits a patent-protected indication or other patent-protected information, then such patents that cover the omitted indication or the omitted information will not be considered to be “applicable to the Biosimilar”. Specifically, we propose at § 429.110(d)(2)(i)(A) through (D) that the Biosimilar Manufacturer must demonstrate that patents related to the Reference Drug are unlikely to prevent the Biosimilar from being marketed before the High Likelihood Deadline through any of four pathways specified. The first option the Biosimilar Manufacturer may demonstrate is that there will be no unexpired patents relating to the reference product included in the Reference Drug that are applicable to the Biosimilar. The second option the Biosimilar Manufacturer may demonstrate is that one or more court decisions or decisions by the United States Patent and Trademark Office (USPTO)'s Patent Trial and Appeal Board (PTAB) establish the invalidity, unenforceability, or non-infringement of any potentially applicable unexpired patents relating to the reference product included in the Reference Drug that a patent holder asserted was applicable to the Biosimilar. The third option the Biosimilar Manufacturer may demonstrate is that neither a court nor PTAB has adversely ruled against the Biosimilar Manufacturer's patent assertion(s) pertaining to unexpired patent(s) relating to the reference product included in the Reference Drug applicable to the Biosimilar, and the Biosimilar Manufacturer has publicly announced a precise launch date for the Biosimilar that is both a calendar date before the High Likelihood Deadline and is not contingent on the outcome of pending litigation. Finally, the fourth option the Biosimilar Manufacturer may demonstrate is that the Biosimilar Manufacturer has a signed agreement with the Reference Manufacturer that permits the Biosimilar Manufacturer to market the Biosimilar before the High Likelihood Deadline, without improper constraints on the Biosimilar Manufacturer. In accordance with the parameters set forth in section 1192(f)(2)(D)(iv) of the Act and proposed § 429.110(c)(1)(iv) of this section, an improper constraint includes, but is not limited to: circumstances in which the Biosimilar Manufacturer is the same as the Reference Manufacturer or is treated as being the same pursuant to section 1192(f)(1)(C) of the Act; an instance in which the Biosimilar Manufacturer has entered into an agreement with the Reference Manufacturer that requires or incentivizes the Biosimilar Manufacturer to submit a Biosimilar Delay Request; and an instance in which a Biosimilar Manufacturer has entered into an agreement with the Reference Manufacturer that directly or indirectly restricts the quantity of the Biosimilar sold in the United States on or after the selected drug publication date of the initial price applicability year for which the Biosimilar Manufacturer is requesting a Biosimilar Delay.
Second, regarding the proposal at § 429.110(d)(2)(ii) that the Biosimilar Delay Request must clearly demonstrate that the Biosimilar Manufacturer will be operationally ready to market the Biosimilar before the High Likelihood Deadline, to assess this requirement, we propose to consider the Biosimilar Manufacturer's progress against the actions, activities, and milestones that are typical of the normal course of business leading up to the marketing of a drug as evidenced by both: (1) disclosures about capital investment, revenue expectations, and actions consistent with the normal course of business for marketing of a biosimilar biological product before the High Likelihood Deadline; and (2) a
( printed page 36250)
manufacturing schedule that is consistent with the public-facing statements and demonstrates readiness to meet revenue expectations. We propose these criteria because we believe they are indicative of operational readiness and should be available in the elements that CMS must consider in making this determination as required by section 1192(f)(1)(B)(ii) of the Act.
In accordance with sections 1192(f)(3)(B), CMS must use information from items described in sections 1192(f)(1)(B)(ii)(I)(bb) and (III) of the Act submitted to CMS by the Biosimilar Manufacturer requesting the Biosimilar Delay to identify if there is clear and convincing evidence that the Biosimilar will be marketed before the High Likelihood Deadline. Consistent with these statutory requirements and the policies implementing section 30.3.1.3 of the Negotiation Program Guidance, we propose at § 429.110(f)(1)(i) through (iii) the information we would review for such “clear and convincing evidence,” which must include—
All agreements related to the Biosimilar filed with the Federal Trade Commission (FTC) or the Assistant Attorney General under subsections (a) and (c) of section 1112 of the Medicare Prescription Drug, Improvement, and Modernization Act of 2003;
To the extent available, the manufacturing schedule for the Biosimilar submitted to FDA during its review of the application for licensure under section 351(k) of the PHS Act for the Biosimilar; and
To the extent available, the Biosimilar Manufacturer's disclosures pertaining to the marketing of the Biosimilar (for example, in filings with the Securities and Exchange Commission required under section 12(b), 12(g), 13(a), or 15(d) of the Securities Exchange Act of 1934 or comparable documentation distributed to the shareholders of privately held companies) about capital investment, revenue expectations, and other actions typically taken by a manufacturer in the normal course of business in the year (or the 2 years, as applicable) before marketing of a Biosimilar.
To illustrate what information specifically that CMS might identify within such documentation to potentially demonstrate that the Biosimilar has a high likelihood of being marketed before the High Likelihood Deadline, we provide three examples of “clear and convincing evidence” that might be included in the documentation required at section 1192(f)(3)(B) of the Act and proposed in § 429.110(f)(1)(i) through (iii). These examples are illustrative but alone may not always constitute “clear and convincing evidence” of a high likelihood of being marketed. First, we provide two examples of evidence that could potentially demonstrate that a patent (or patents) related to the Reference Drug are unlikely to prevent the Biosimilar from being marketed: (1) the listing of a signed agreement between the Biosimilar and Reference Drug Manufacturers under “Legal Proceedings” or another section, as appropriate, in a Form 10-K, along with a copy of the agreement if required to be filed with the Federal Trade Commission (FTC) or the Assistant Attorney General under the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, and (2) the lack of any adverse actions from a court or PTAB under “Legal Proceedings” or another section, as appropriate, in a Form 10-K pertaining to a Biosimilar Manufacturer's patent assertion(s) of an unexpired patent or patent(s) relating to the reference product included in the Reference Drug applicable to the Biosimilar, and the Biosimilar Manufacturer publicly announced a precise launch date for the Biosimilar by a calendar date prior to the High Likelihood Deadline within the operational preparations and/or other steps to market and/or produce the Biosimilar under “Management's Discussion and Analysis (MD&A)” or another section, as appropriate, in a Form 10-K. Second, we provide one example of evidence that could potentially demonstrate that the Biosimilar Manufacturer is operationally ready: information regarding the operational preparations and/or other steps to market and/or produce the Biosimilar under “MD&A” or another section, as appropriate, in a Form 10-K. These distinct examples are intended to be for illustrative purposes only and do not supersede the requirements of the originating authorities for the required documentation (for example, the Securities Exchange Act of 1934 governing disclosure requirements). Consistent with section 1192(f)(1)(B)(ii)(III)(bb) of the Act, comparable documentation that is distributed to the shareholders of privately held companies could be provided in lieu of any examples of disclosures required under the Securities Exchange Act of 1934 for publicly traded companies. These examples are not exhaustive of the information that might be included in the documentation required at section 1192(f)(3)(B) of the Act and in a submission for a request for an Initial Delay Request necessary to demonstrate “clear and convincing evidence”.
Finally, consistent with section 1192(f)(1)(B)(ii)(II) of the Act and at proposed § 429.110(f)(2), we may request additional information from the Biosimilar Manufacturer as necessary to make a determination with respect to the Initial Delay Request after reviewing an Initial Delay Request. Any such written request would specify the additional information required, a form and manner in which the Biosimilar Manufacturer must provide the additional information, and the deadline for providing such information.
As proposed at § 429.110(g)(1), we would provide in writing a notice of determination, on or after the selected drug publication date for the initial price applicability year by a specific date to be set forth by CMS, to the Biosimilar Manufacturer that requested the Initial Delay Request regarding whether the request was successful or unsuccessful. If unsuccessful, we would specify the reason for the unsuccessful request. Such reasons provided may include: (1) failure to submit all elements of the Biosimilar Delay Request by the applicable deadline (CMS-10844, OMB 0938-1443); (2) failure to meet another statutory requirement for granting a request (other than the high likelihood requirement), including in the case that the Reference Drug would not have been a selected drug for the initial price applicability year absent the Initial Delay Request; or (3) failure to demonstrate a high likelihood that the Biosimilar will be licensed and marketed before the High Likelihood Deadline. We also propose at § 429.110(g)(1)(i)(B) to notify each Reference Manufacturer named in a successful Biosimilar Delay Request. We propose that such notification would be in writing and would identify the Reference Drug that would have been a selected drug in the initial price applicability year, absent the successful Initial Delay Request. Reference Manufacturers named in unsuccessful Initial Delay Requests would not be notified. We will publish the number of Reference Drugs that would have been selected drugs for the initial price applicability year, absent successful Initial Delay Requests, as part of publishing the selected drug list as proposed in § 429.100 and described in section II.B.1. of this proposed rule (see proposed § 429.110(g)(2)).
Section 1192(f)(2)(B) of the Act requires CMS to determine whether each Biosimilar named in a successful Initial Delay Request is licensed and
( printed page 36251)
marketed during the Initial Delay Period. CMS proposes at § 429.110(h)(1) that we would determine whether each Biosimilar named in a successful Initial Delay Request was licensed and marketed during the Initial Delay Period. If we determine that the Biosimilar is not licensed and marketed during the Initial Delay Period, we propose at § 429.110(h)(1)(i) that the Biosimilar Manufacturer will have the opportunity to submit an Additional Delay Request. In proposed § 429.110(g)(3), for successful Initial Delay Requests submitted with respect to the initial price applicability year, we propose to notify a Biosimilar Manufacturer if CMS has determined that the Biosimilar named in the Biosimilar Manufacturer's successful Initial Delay Request is licensed and marketed during the Initial Delay Period by a date to be specified by CMS in technical guidance, which will be no later than the end of October of the calendar year of the selected drug publication date for the initial price applicability year for which the Biosimilar Manufacturer submitted the successful Initial Delay Request. For example, if CMS determined that a Biosimilar Manufacturer's Initial Delay Request was successful for initial price applicability year 2029, CMS would provide this notification to the Biosimilar Manufacturer no later than the end of October 2027.
If the Biosimilar Manufacturer chooses to submit an Additional Delay Request, sections 1192(f)(2)(B)(i)(I) and (iii) include requirements for an Additional Delay Request. We propose these requirements in § 429.110(e), along with the corresponding documentation requirements in § 429.110(f). Consistent with section 1192(f)(2) of the Act, to first be eligible for an Additional Delay Request, we would need to determine that the Biosimilar listed in the Additional Delay Request was identified in a successfully granted Initial Delay Request (consistent with proposed § 429.110(c)) and the licensure and marketing under section 351(k) of the PHS Act has not commenced between the publication date of the selected drug list for the initial price applicability year for which the Initial Delay Request was granted and the date that is 1 year following that publication date. We propose these requirements at § 429.110(e)(1)(i) and (ii). Additionally, as a threshold requirement, we would determine that the requirements proposed at § 429.110(c)(1)(ii) through (iv) for an Initial Delay Request, in accordance with sections 1192(f)(1) and (2) of the Act, remain met for purposes of the Additional Delay Request (see proposed § 429.110(e)(1)(iii)). Further, in accordance with section 1192(f)(2)(D)(ii) of the Act and as described in proposed § 429.110(e)(1)(iv), a Biosimilar named in the Biosimilar Manufacturer's successful Initial Delay Request is not eligible for an Additional Delay Request if the status of the Reference Drug would change to a long-monopoly drug (as defined in proposed § 429.20), with respect to the initial price applicability year for which the Biosimilar Manufacturer is submitting an Additional Delay Request. If the requirements proposed in § 429.110(e)(1)(i) through (iv) are met, we would then reevaluate and determine whether the requirements in proposed § 429.110(d) regarding whether there is a high likelihood that the Biosimilar will be licensed and marketed before the High Likelihood Deadline continue to be met as proposed in § 429.110(e)(2). Finally, in accordance with section 1192(f)(2)(B)(i)(II) of the Act and as described in proposed § 429.110(e)(3), we must determine, on the basis of clear and convincing evidence, that the Biosimilar Manufacturer has made a significant amount of progress towards both licensure and marketing of the Biosimilar since the Biosimilar Manufacturer's submission of the successful Initial Delay Request. In accordance with section 1192(f)(2)(B)(i)(II) of the Act, CMS is required to use information from the following items when assessing whether there is clear and convincing evidence that the Biosimilar Manufacturer has made a significant amount of progress towards licensure and marketing of the Biosimilar since the Biosimilar Manufacturer's submission of the successful Initial Delay Request for the Biosimilar: (1) all agreements related to the Biosimilar filed with the FTC or the Assistant Attorney General pursuant to subsections (a) and (c) of section 1112 of the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (as described in section 1192(f)(1)(B)(ii)(I)(bb) of the Act); and (2) additional information and documents that CMS may request after CMS has reviewed the information required for submission of the Additional Delay Request necessary to make a determination about an Additional Delay Request (as described in section 1192(f)(1)(B)(ii)(II) of the Act).
Consistent with implementation of policies in section 30.3.1.4 of the Negotiation Program Guidance, recognizing that approximately 1 year has passed since submission of the successful Initial Delay Request, we would consider whether the Biosimilar Manufacturer demonstrates that the Biosimilar will be licensed and marketed before the High Likelihood Deadline. Specifically, we propose at § 429.110(e)(3) that the determination of whether a significant amount of progress has been made by the Biosimilar Manufacturer towards licensure and marketing of the Biosimilar since the successful Initial Delay Request submission for such Biosimilar will be based on a holistic review of the documentation submitted with the Additional Delay Request (as described in proposed § 429.110(f)(1), including any follow-up documentation requests from CMS to the manufacturer described in proposed § 429.110(f)(2)). Within the request we would consider if the Biosimilar Manufacturer can demonstrate affirmative progress towards being operationally ready to market the Biosimilar, meaning that we would consider the Biosimilar Manufacturer's progress on the actions, activities, and milestones that are typical of the normal course of business leading up to the marketing of a drug since the successful Initial Delay Request submission for the Biosimilar evidenced in any updates or supplements to the documents specified in section 1192(f)(1)(B)(ii)(III) of the Act and as described in proposed § 429.110(f)(1). Additionally, we would consider if the manufacturing schedule (as provided in § 429.110(f)(1)(ii)) is consistent with the public-facing statements (that may be identified within the information provided in the materials set forth at proposed § 429.110(f)(1)(iii)) and demonstrates readiness to meet revenue expectations.
After completing our review of an Additional Delay Request, similar to the process for notification after an Initial Delay Request, we would notify the Biosimilar Manufacturer that submitted the Additional Delay Request regarding CMS' determination of whether the Additional Delay Request was successful or unsuccessful (see proposed § 429.110(g)(1)(i)(A)). We also propose to notify the Reference Manufacturer of a successful Additional Delay Request (see proposed § 429.110(g)(1)(i)(B)) and would publish the number of Reference Drugs that would have been selected drugs for the initial price applicability year if they had not been determined eligible by CMS for a Biosimilar Delay Request for that initial price applicability year (see proposed § 429.110(g)(2)).
( printed page 36252)
(c) Review For Failure of the Biosimilar To be Licensed and Marketed; Rebate Owed for Failure of a Biosimilar To be Licensed and Marketed (§ 429.110(h) through (i))
As discussed previously, CMS proposes at § 429.110(h)(1) that we would determine whether each Biosimilar named in a successful Initial Delay Request was licensed and marketed during the Initial Delay Period. If we determine that the Biosimilar is not licensed and marketed during the Initial Delay Period, we propose at § 429.110(h)(1)(i) that the Biosimilar Manufacturer will have the opportunity to submit an Additional Delay Request. In proposed § 429.110(h)(1)(ii), we propose that if the Biosimilar Manufacturer chooses not to submit an Additional Delay Request, or submits an Additional Delay Request that CMS determines does not meet all requirements in proposed § 429.110(e), CMS would include the Reference Drug on the selected drug list for the initial price applicability year that is 1 year after the initial price applicability year for which the Reference Drug would have been included on the selected drug list if not for the successful Initial Delay Request (for example, the selected drug list for initial price applicability year 2030 for successful Initial Delay Requests for initial price applicability year 2029), unless a different biosimilar biological product is marketed before the publication of the selected drug list for the applicable initial price applicability year, in which case CMS could also determine, in accordance with section 1192(c) of the Act and described in sections II.B.6.d. of this proposed rule, that the Reference Drug no longer meets the criteria to be a selected drug and will be excluded from such applicable list of drugs selected for an initial price applicability year. Further, in accordance with section 1192(f)(2)(C) of the Act and as described in proposed § 429.110(h)(2), CMS must determine whether each Biosimilar named in a successful Additional Delay Request is licensed and marketed during the Second Delay Period. We propose at § 429.110(h)(2)(i) that if CMS determines that the Biosimilar is not licensed and marketed during the Second Delay Period, unless a different biosimilar biological product is marketed, CMS would include the Reference Drug on the selected drug list for the initial price applicability year that is 2 years after the initial price applicability year for which the Reference Drug would have been included on the selected drug list if not for the successful Initial Delay Request.
In accordance with sections 1192(f)(2)(B)(ii), 1192(f)(2)(C), and 1192(f)(4)(A) of the Act and as described in proposed § 429.110(i)(1), if (1) CMS delayed the selection and negotiation of a Reference Drug for 1 or 2 years, (2) CMS determined that the Biosimilar was not licensed and marketed, and (3) the manufacturer of the Reference Drug agrees to an MFP for the Reference Drug, the Reference Manufacturer is required to pay a rebate for the years that the manufacturer would have provided access to the MFP for the Reference Drug but for the delay. In accordance with section 1192(f)(4)(B) of the Act, we specify in proposed § 429.110(i)(4) that the rebate owed by the Reference Manufacturer, for the year for which an Initial Delay Request and, if applicable, an Additional Delay Request was granted will be calculated as follows:
In accordance with section 1192(f)(4)(B)(i) of the Act and as described in proposed § 429.110(i)(4)(ii), in the case of a Reference Drug that is a drug covered under Part D, 75 percent of the difference between the AMP, with respect to each of the calendar quarters of the price applicability period, and the MFP negotiated for the Reference Drug multiplied by the number of units dispensed under Part D for the Reference Drug in each calendar quarter of the price applicability period that would have applied but for the delay.
In accordance with section 1192(f)(4)(B)(ii) of the Act and as described in proposed § 429.110(i)(4)(iii), in the case of a Reference Drug payable under Part B, 80 percent of the difference between the payment amount under section 1847A(b) of the Act, with respect to each of the calendar quarters of the price applicability period, and the MFP negotiated for the Reference Drug, multiplied by the number of units of the billing and payment code of the Reference Drug administered or furnished under Part B (excluding units that are packaged into the payment amount for an item or service and are not separately payable under Part B) for each calendar quarter of the price applicability period that would have applied but for the delay.
As described in proposed § 429.110(i)(4)(iv), in the case of a Reference Drug that is a drug covered under Part D and payable under Part B, the rebate amount will be calculated by summing the rebate amount for the units payable under Part B as specified in proposed § 429.110(i)(4)(iii) and the rebate amount for units covered under Part D as specified in proposed § 429.110(i)(4)(ii).
For the year for which an Additional Delay Request was granted, we will adjust the MFP as described in section 1195(b)(1)(A) of the Act to account for changes in the CPI-U. Additionally, before applying a rebate as described in proposed § 429.110(i)(5), we will determine if the Reference Drug transitioned to a long monopoly drug, at the time of its inclusion on the selected drug list for the initial price applicability year. For drugs payable under Part B and covered under Part D, we would calculate the rebate for the units payable under Part B following the Part B formula and we would calculate the rebate for the units covered under Part D following the Part D formula.
In the case of a Reference Drug that CMS determines transitioned to a long-monopoly drug during the delay, in accordance with section 1192(f)(4)(C) of the Act and as described in proposed § 429.110(i)(5) through (6), the rebate calculation will substitute the MFP negotiated for the Reference Drug with the following amount. The amount will be equal to 65 percent of the average non-FAMP (consistent with proposed § 429.20 and defined in 38 U.S.C. 8126(h)(5)) for 2021 (or the first full year following market entry if there is no non-FAMP for 2021) increased by the percentage increase in the CPI-U from September 2021 (or December of such first full year following the market entry) to September of the year prior to the selected drug publication date for the initial price applicability year that would have applied but for the Initial Delay Request. For example, if inclusion of the Reference Drug on the selected drug list is delayed until initial price applicability year 2030 due to a successful Initial Delay Request, and the Reference Drug transitions to a long-monopoly drug, the rebate calculation will use September of the year prior to the selected drug publication date for initial price applicability year 2029 (September 2026) for the purposes of adjusting for inflation the average non-FAMP for 2021. As described in proposed § 429.110(i)(6), the rebate calculation will substitute the MFP negotiated for the Reference Drug with the amount that is further adjusted by the annual percentage increase in the CPI-U for the 12-month period ending with July of the calendar year that is 2 years before the initial price applicability year for which the Additional Delay Request was granted.
In accordance with section 1192(f)(4)(B) of the Act and as described in proposed § 429.110(i)(4)(i), we intend to apply the MFP to the rebate calculation for all the previous initial applicability years where the Reference
( printed page 36253)
Drug would have been on the selected drug list if not for the successful Biosimilar Delay Request. For example, if the Reference Drug would have been on the list for initial price applicability years 2029 and 2030 but for the approval of an Initial Delay Request and an Additional Delay Request, and CMS determines the Biosimilar was not licensed and marketed, we will use the MFP agreed to for initial price applicability year 2031 to calculate the rebate amount for initial price applicability years 2029 and 2030.
In accordance with section 1192(f)(4)(D) of the Act and as described in proposed § 429.110(i)(3), the rebates paid for drugs payable under Part B would be deposited in the Federal Supplementary Medical Insurance Trust Fund established under section 1841 of the Act. The rebates paid for drugs covered under Part D would be deposited in the Medicare Prescription Drug Account established under section 1860D-16 of the Act, which is within the Federal Supplementary Medical Insurance Trust Fund. Under proposed § 429.110(i)(2), we would specify a form and manner for the administration of rebates, including the timing and mechanism for notifying manufacturers when a rebate is owed and the process for payment, in future rulemaking.
4. Identification of Negotiation-Eligible Drugs (§ 429.115)
Section 1192(d)(1) of the Act requires that a “negotiation-eligible drug” means, with respect to the selected drug publication date with respect to an initial price applicability year, a qualifying single source drug, as defined in section 1192(e) of the Act, that is either a Part D high spend drug or a Part B high spend drug. Section 1192(d)(1)(A) of the Act describes a Part D high spend drug as a qualifying single source drug that is among the 50 qualifying single source drugs with the highest total expenditures under part D of Title XVIII, as determined by the Secretary in accordance with section 1192(d)(3) of the Act, during the most recent 12-month period for which data are available prior to such selected drug publication date (but ending no later than October 31 of the year prior to the year of such drug publication date). Section 1192(d)(1)(B) of the Act describes a Part B high spend drug as a qualifying single source drug that is among the 50 qualifying single source drugs with the highest total expenditures under part B of Title XVIII, as determined by the Secretary in accordance with section 1192(d)(3) of the Act, during such most recent 12-month period describes in section 1192(d)(1)(A) of the Act. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 30.2 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
We are proposing to codify the statutory requirements in section 1192(d) of the Act at § 429.115, including that a negotiation-eligible drug for an initial price applicability year is a qualifying single source drug, as identified under proposed § 429.125, that is among the 50 qualifying single source drugs with the highest total expenditures under Part D, or among the 50 qualifying single source drugs with the highest total expenditures under Part B. We are proposing to codify our process for identifying the negotiation-eligible drugs for each initial price applicability year, consistent with the process implemented through prior guidance, as follows.
We propose at § 429.115(a)(1) to identify Part D high spend drugs described in section 1192(d)(1)(A) of the Act using the following steps. We would first remove from negotiation eligibility any qualifying single source drugs that are already selected drugs in accordance with section 1192(d)(3)(A)(i) of the Act. Next, for remaining qualifying single source drugs, CMS would calculate a qualifying single source drug's total expenditures under Part D using the methodology set forth at proposed § 429.120(a) and described in section II.B.5. of this proposed rule and rank those qualifying single source drugs by total expenditures under Part D during the total expenditures measurement period. Finally, we would identify the 50 qualifying single source drugs that have the highest total expenditures under Part D during the total expenditures measurement period (that is, Part D high spend drugs).
Then, we are proposing at § 429.115(a)(2) to identify Part B high spend drugs described in section 1192(d)(1)(B) of the Act using the following steps. As with Part D high spend drugs, we would first remove from negotiation eligibility any qualifying single source drugs that are already selected drugs in accordance with section 1192(d)(3)(A)(i) of the Act.[18]
Next, for remaining qualifying single source drugs, CMS would calculate a qualifying single source drug's total expenditures under Part B using the methodology set forth at proposed § 429.120(b) and described in section II.B.5. of this proposed rule and rank the remaining qualifying single source drugs by total expenditures under Part B during the total expenditures measurement period. Finally, we would identify the 50 qualifying single source drugs that have the highest total expenditures under Part B during the total expenditures measurement period (that is, Part B high spend drugs).
We are proposing at § 429.115(b) that, when identifying Part D high spend drugs and Part B high spend drugs as proposed at § 429.115(a)(1)(iv) and (a)(2)(iv), respectively, if two or more qualifying single source drugs have the same total expenditures to the cent under Part D or Part B, and such total expenditures are the 50th highest among qualifying single source drugs under Part D or Part B, we would rank the qualifying single source drugs based on which drug has the earlier approval or licensure date, as applicable, associated with the earliest-approved FDA application belonging to the NDA/BLA holder and containing the active moiety/active ingredient in the drug, until we have identified 50 Part D high spend drugs and Part B high spend drugs, respectively. These 50 Part D high spend drugs and 50 Part B high spend drugs, identified in accordance with proposed § 429.115(a)(1) and (a)(2), respectively, would be the negotiation-eligible drugs for the initial price applicability year. This proposal is a modification from Negotiation Program Guidance, which established that for initial price applicability years 2026, 2027, and 2028, we would identify qualifying single source drugs with the same total expenditures to the dollar. As noted in section II.B.2. of this proposed rule, we believe that using information to the cent, rather than to the dollar as under prior policy, is more precise for purposes of determining negotiation-eligible drugs.
5. Calculation of Total Expenditures (§ 429.120)
As described in sections II.B.2., II.B.4., and II.B.6.c.2. of this proposed rule, we are proposing at §§ 429.105(a), 429.115(a), and 429.125(e)(2) to calculate total expenditures under Part B and Part D as a step in the processes for identifying selected drugs, negotiation-eligible drugs, and drugs eligible for the low-spend Medicare drug exclusion, respectively. Section 1191(c)(5) of the Act defines the term “total expenditures” to include, in the case of expenditures with respect to Part D, the total gross covered prescription
( printed page 36254)
drug costs (as defined in section 1860D-15(b)(3) of the Act). In the case of “total expenditures” with respect to Part B, section 1191(c)(5) of the Act specifies that such term excludes expenditures for a drug or biological product that are bundled or packaged into the payment for another service. With respect to initial price applicability years 2026 through 2028, we explained through guidance how we will implement the statutory requirement to calculate total expenditures under Part B and total expenditures under Part D, including, for example, section 30 of the Negotiation Program Guidance with respect to initial price applicability year 2028. We are proposing to codify the definition of total expenditures in section 1191(c)(5) of the Act at § 429.20, and we propose how we would calculate total expenditures under Part B and Part D at § 429.120.
a. Calculation of Total Expenditures Under Part D
At § 429.120(a), we propose to calculate total expenditures under Part D for a given potential qualifying single source drug, qualifying single source drug, negotiation-eligible drug, or selected drug, as the sum of gross covered prescription drug costs for each PDE record for such drug that meets the criteria in proposed § 429.120(a)(1) through (a)(5). CMS would identify these PDE records as follows: (1) the dates of service are during the total expenditures measurement period (to allow a reasonable time for Part D plan sponsors to submit PDE data, we would use PDE data for the dates of service in the total expenditures measurement period that are available in CMS' data repository by the November 30 following the total expenditures measurement period (or the first business day following November 30 if November 30 does not fall on a business day)); (2) total gross covered prescription drug costs on the PDE record is greater than zero dollars; (3) the PDE record is considered final action; [19]
(4) the drug coverage status code indicates the PDE record is for a drug covered under Part D; and (5) the compound code indicates the PDE record is not for a compounded drug.[20]
b. Calculation of Total Expenditures Under Part B
At § 429.120(b), we propose a methodology for calculating total expenditures under Part B for a given potential qualifying single source drug, qualifying single source drug, negotiation-eligible drug, or selected drug. This methodology would use a combination of total allowed charges from Original Medicare (OM) Part B claims data (inclusive of beneficiary coinsurance and Medicare payment) and a comparable amount calculated using Medicare Advantage (MA) encounter data for Part B items and services, which would reflect the amount that would have been applicable under OM. Then, we would sum total expenditures under Part B based on OM Part B claims data and total expenditures under Part B for MA encounter data. To allow a reasonable time for providers and suppliers to submit OM Part B claims data and Medicare Advantage Organizations to submit MA encounter data for Part B items and services, we would use Part B data for the dates of service in the total expenditures measurement period that are available in CMS' data repository by November 30 following the total expenditures measurement period (or the first business day following November 30 if November 30 does not fall on a business day).
We received many comments on the draft guidance for initial price applicability year 2028 and manufacturer effectuation of the MFP in 2026, 2027, and 2028 suggesting that CMS should account for expenditures on drugs payable under Part B and administered to MA enrollees when identifying Part B high spend drugs. In response to these comments, we stated in the Negotiation Program Guidance that we agreed with these commenters that MA expenditures for such drugs should be accounted for and included in the calculation of total expenditures under Part B, and we described CMS' methodology, consistent with the previous paragraph, for including such expenditures in the calculation of total expenditures under Part B. In this proposed rule, we reiterate and expand upon the discussion in the Negotiation Program Guidance.
More than half (54 percent [21]
) of Medicare enrollees were enrolled in MA plans in 2025. We would therefore exclude a significant portion of total spending on drugs payable under Medicare Part B by only using Part B claims data in the calculation of total expenditures under Part B. Such an approach would skew the negotiation-eligible drug list toward drugs with high expenditures under Part D and away from drugs with high expenditures under Part B and therefore could misrepresent the highest spend drugs. There is no indication that statute intends the negotiation-eligible drug list to skew towards drugs with high expenditures under Part D; rather, section 1192(d)(1) of the Act indicates equal treatment of drugs with high expenditures under Part D and drugs with high expenditures under Part B, requiring CMS to identify 50 Part D high spend drugs and 50 Part B high spend drugs beginning in initial price applicability year 2028.
Consistent with the policy adopted in Negotiation Program Guidance, in this proposed rule we propose that the term “total expenditures under part B of Title XVIII” as defined at section 1191(c)(5) of the Act and as used in the Negotiation Program, is best read to include MA expenditures for drugs payable under Part B and administered to MA enrollees. In the case of expenditures with respect to Part B, section 1191(c)(5) of the Act provides only that the term “total expenditures” excludes expenditures for a drug or biological product that are bundled or packaged into the payment for another service.
Statutory language in Title XVIII of the Act and sections 11001 and 11002 of the IRA suggest MA expenditures ought to be included in “total expenditures under part B of title XVIII” for purposes of the Negotiation Program. First, section 1852(a)(1) of the Act requires MA plans to provide to enrollees the “benefits under the original [M]edicare [Fee-For-Service]
( printed page 36255)
program option,” including, as relevant here, drugs payable under Part B. For purposes of determining “total expenditures” with respect to Part B for purposes of the Negotiation Program, we believe that MA expenditures for drugs payable under Part B may thus be understood as expenditures provided under this requirement to provide benefits available under Part B, and appropriately included in total expenditures under Part B for such drugs.
Further, section 1191(c)(2)(B) of the Act requires that, for purposes of the Negotiation Program, a “maximum fair price eligible individual” includes “in the case such drug is furnished or administered to the individual by a hospital, physician, or other provider of services or supplier, an individual who is enrolled under part B of title XVIII, including an individual who is enrolled in an MA plan under part C of such title, if payment may be made under part B for such selected drug.” Including MA expenditures in the definition of total expenditures under Part B is consistent with the statutory approach reflected in this definition, which considers “individual[s] enrolled in an MA plan” to be “include[ed]” within the reference to individuals “enrolled under part B of Title XVIII” to the extent “payment may be made under part B” for a selected drug.
Finally, section 1191(c)(5) of the Act's definition of total expenditures under Part B identifies explicitly one exclusion—expenditures where payment for the drug is bundled with payment for another Part B service—but does not similarly exclude MA expenditures. As noted previously, the exclusion of MA expenditures would result in far more significant consequences for the identification of negotiation-eligible and selected drugs under the Negotiation Program than the exclusion that is identified explicitly. In light of the statutory indicia favoring inclusion of MA expenditures discussed previously and the significant consequences with respect to the Negotiation Program should MA expenditures be excluded, we believe the absence of clear statutory language excluding such expenditures weighs in favor of including MA expenditures in the definition of total expenditures under Part B.
For these reasons, we are proposing at § 429.120(b) a methodology to include MA expenditures in the calculation of total expenditures under Part B. As we noted in the Negotiation Program Guidance, MA encounter data for Part B items and services does not reliably include the actual amount paid by the MA plan sponsor. Due to this gap in MA encounter data for Part B items and services, we believe it appropriate to estimate MA expenditures for drugs payable under Part B by using MA encounter data for Part B items and services to identify the units of drugs payable under Part B that were administered under MA and then determining what Medicare would have paid for such units under OM Part B. Accordingly, we propose to use the following methodology to calculate total expenditures under Part B:
Total expenditures under Part B based on OM Part B claims data
would equal the sum of the total allowed charges for each OM Part B claim for a qualifying single source drug that meets the following criteria: (1) date of service is during the total expenditures measurement period; (2) the claim type is associated with an OM Part B claim in an outpatient setting (including but not limited to clinics, Federally Qualified Health Centers, and ambulatory surgical centers), a professional services claim, or durable medical equipment claim (currently, these claim type codes are 40, 71, 72, 81, or 82); (3) the total allowed charges (defined as the amount that is inclusive of the beneficiary coinsurance and Medicare payment for covered Part B items and services) for the claim line is greater than $0; (4) the claim is considered final action; [22]
(5) the claim is not billed as a compounded drug; and (6) the claim is not for a drug or biological product that is bundled or packaged into the payment for another service under Part B OM. We have identified rare instances where claims for separate payment have been submitted for drugs payable under Part B when such claims are typically payable only as part of a bundled payment. We are proposing to exclude such separately billed claims.
Total expenditures under Part B based on MA encounter data for Part B items
would equal the sum of the total allowed charges that would have been applicable under OM Part B for each MA encounter data record for Part B services for such drug that meets the following criteria: (1) date of service is during the total expenditures measurement period; (2) the claim type is associated with an MA encounter record in an outpatient setting (including but not limited to clinics, Federally qualified health centers, and ambulatory surgical centers), professional services, or durable medical equipment record, as determined by CMS (currently, these claim types are 4012, 4013, 4014, 4022, 4023, 4032, 4034, 4071, 4072, 4073, 4074, 4075, 4076, 4077, 4079, 4083, 4085, 4087, 4089, 4700, and 4800); (3) the reported total number of units on the MA encounter data record line is greater than zero; (4) the encounter data record is considered final action; [23]
(5) the encounter data record is not denied; (6) the encounter data record is not a chart review record; (7) the encounter data record line is not for a supplemental benefit; (8) the encounter data record is not reported as a compounded drug; and (9) the encounter data record is not for a drug or biological product that is bundled or packaged into the payment for another service under Part B OM. In instances where an encounter data record for separate payment is submitted for a drug payable under Part B when such a claim is typically payable under Part B OM payment rules only as part of a bundled payment, such claim will be considered to be bundled or packaged into the payment for another service and will not be included in the total allowed charges calculation. To calculate the total allowed charges that would have been applicable under OM Part B for each of the aforementioned MA encounter data records, we would first adjust the unit field in MA encounter data for Part B items and services by referencing the Medically Unlikely Edits (MUEs), which are designed to reduce improper payments.[24]
Because Medicare Administrative Contractors apply these edits to OM Part B claims, this would bring the MA encounter data for Part B items and services into closer alignment. We would then multiply the adjusted units by the appropriate published payment limit (for example, Average Sales Price (ASP)-based) or payment rate (for example, Outpatient Prospective Payment System (OPPS), Ambulatory Surgical Center (ASC)) to calculate what would have been applicable for the Part B items and services under OM.
Typically, “single source drugs and biologicals” as defined in section 1847A(c)(6)(D) of the Act are assigned to unique HCPCS codes; however, there may be cases where a qualifying single source drug is assigned to a HCPCS code with other products. In such cases, we are proposing to use the apportionment
( printed page 36256)
methodology proposed in § 429.120(b)(3) wherein CMS would use ASP sales volume data to apportion Part B total expenditures based on the ratio of reported sales volume of the qualifying single source drug compared to reported sales volume of all products assigned to the HCPCS code to calculate the total expenditures under Part B.
6. Identification of Qualifying Single Source Drugs (§ 429.125)
Section 1192(e)(1) of the Act requires that the term “qualifying single source drugs” means, with respect to an initial price applicability year, subject to sections 1192(e)(2) through 1192(e)(4) of the Act, a covered part D drug (as defined in section 1860D-2(e) of the Act) that is described in section 1192(e)(1)(A) or section 1192(e)(1)(B) of the Act, or a drug or biological product for which payment may be made under part B of title XVIII that is described in section 1192(e)(1)(A) or section 1192(e)(1)(B) of the Act. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example section 30.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
We are proposing that, with respect to each initial price applicability year, a qualifying single source drug is a drug covered under Part D, a drug payable under Part B, or both, as such terms are defined at proposed § 429.20, that meets the statutory criteria set forth in section 1192(e) of the Act. Specifically, we propose in § 429.125(a)(1) to codify the requirements in section 1192(e)(1)(A) of that Act that, for drug products, a qualifying single source drug is a drug covered under Part D, payable under Part B, or both: (1) that is approved under section 505(c) of the FD&C Act and marketed pursuant to such approval; (2) for which, as of the selected drug publication date with respect to a given initial price applicability year, at least 7 years have elapsed since the date of such approval; and (3) that is not the listed drug for any drug approved and marketed under an Abbreviated New Drug Application (ANDA) under section 505(j) of the FD&C Act. We propose in § 429.125(a)(2) to codify the requirements in section 1192(e)(1)(B) of the Act that, for biological products, a qualifying single source drug is a drug covered under Part D, payable under Part B, or both: (1) that is licensed under section 351(a) of the Public Health Service Act (“PHS Act”) and marketed pursuant to such licensure; (2) for which, as of the selected drug publication date with respect to a given initial price applicability year, at least 11 years have elapsed since the date of such licensure; and (3) that is not the reference product for any biological product that is licensed and marketed under section 351(k) of the PHS Act.
a. Identification of Potential Qualifying Single Source Drugs (§ 429.125(b))
To identify drugs or biological products for purposes of identifying qualifying single source drugs that meet the criteria in section 1192(e) of the Act, we propose to identify drugs and biological products that are potential qualifying single source drugs as described at proposed § 429.125(b).
Sections 11001 and 11002 of the IRA do not define what a “drug” or “biological product” is for purposes of identifying whether such a drug or biological product is a qualifying single source drug. However, the Act provides that a drug or biological product may have multiple dosage forms, strengths, formulations, package sizes, or package types, and multiple applications and approvals. Specifically, for purposes of determining whether a qualifying single source drug is a negotiation-eligible drug under section 1192(d)(1) of the Act, section 1192(d)(3)(B) of the Act states that CMS shall use data that is aggregated across dosage forms and strengths of the drug, including new formulations of the drug, such as an extended release formulation, and not based on the specific formulation, package size, or package type of the drug. Likewise, section 1192(d)(3)(B) of the Act's aggregation rule applies to the calculation of a drug or biological product's total expenditures for purposes of determining whether such drug or biological product meets the low-spend Medicare drug exclusion from a qualifying single source drug, described in section 1192(e)(3)(B) of the Act. Similarly, section 1196(a)(2) of the Act directs CMS to establish procedures “to compute and apply the MFP different strengths and dosage forms of a selected drug and not based on the specific formulation or package size or package type of such drug.” In addition, section 1194(e)(1)(D) of the Act instructs CMS, for purposes of the negotiation process (discussed in further detail in section II.F. of this proposed rule), to consider, among other information, “applications and approvals under section 505(c) of the Federal Food, Drug, and Cosmetic Act or section 351(a) of the Public Health Service Act,” in the plural, for the “drug,” in the singular.
Different dosage forms and strengths, as well as different formulations, of a drug or biological product, containing the same active moiety/active ingredient, may be approved or licensed in multiple NDAs or BLAs. Defining a potential qualifying single source drug on the basis of a single NDA/BLA, and thereby excluding from such potential qualifying single source drug dosage forms and strengths and new formulations of the drug or biological product approved or licensed under other NDAs/BLAs, would be inconsistent with these statutory provisions. To give full effect to all relevant provisions of the statute, including sections 1192(d)(3)(B), 1192(e), 1194(e)(1)(D), and 1196(a)(2) of the Act, we are proposing at § 429.125(b) a process, consistent with policies for implementation as described in, for example, section 30.1 of the Negotiation Program Guidance subject to proposed modifications as noted herein, to identify a potential qualifying single source drug, for purposes of identifying qualifying single source drugs that meet the statutory criteria under section 1192(e) of the Act, using the specific constituent dosage forms and strengths (at the NDC-9 or NDC-11 level) that are identified as aggregated under the same NDA/BLA holder for the same active moiety/active ingredient.[25]
The policies proposed in § 429.125(b), like the policies in, for example, section 30.1 of the Negotiation Program Guidance, would address how CMS is interpreting the statutory directive in section 1192(e) of the Act to identify “drug[s]” or “biological product[s]” for purposes of evaluating whether such drug or biological product satisfies the criteria for qualifying single source drugs.
For drugs, we are proposing at § 429.125(b)(1) to identify a potential qualifying single source drug using all dosage forms and strengths of the drug with the same active moiety and the same holder of an NDA, inclusive of products that are marketed pursuant to different NDAs. If there are multiple NDAs with the same active moiety that include non-identical names reported for the NDA holder, including situations where it appears the NDA holder name has not yet been updated, we are proposing that we may further investigate whether such NDA(s) are held by the same entity for the purposes
( printed page 36257)
of identifying a potential qualifying single source drug using FDA sources as well as relevant publicly available information as CMS deems appropriate. The potential qualifying single source drug would also include all dosage forms and strengths of the drug with the same active moiety and marketed pursuant to the same NDA(s) described in the prior sentences that are: (1) repackaged and relabeled products (defined at proposed § 429.20 to be consistent with 21 CFR 207.1) that are marketed pursuant to such NDA(s); (2) authorized generic drugs (defined in section 1192(e)(2)(B)(i) of the Act and at proposed § 429.20 and described further later in this section) that are marketed pursuant to such NDA(s); or (3) multi-market approval (MMA) [26]
products imported under section 801(d)(1)(B) of the FD&C Act that are marketed pursuant to such NDA(s). Any dosage forms and strengths of the drug with the same active moiety that are distributed by a private label distributor and marketed pursuant to such NDAs would also be aggregated in the potential qualifying single source drug of that NDA holder consistent with the policies for implementation as described in, for example, section 30.1 of the Negotiation Program Guidance.
For biological products, we are proposing at § 429.125(b)(2) to identify a potential qualifying single source drug using all dosage forms and strengths of the biological product with the same active ingredient and the same holder of a BLA, inclusive of products that are marketed pursuant to different BLAs. If there are multiple BLAs with the same active ingredient (or the same antigen component for a biological product that is a vaccine for infectious disease(s), as further described later in this section) that include non-identical names reported for the BLA holder, including situations where it appears the BLA holder name has not yet been updated, we are proposing that we may further investigate whether such BLA(s) are held by the same entity for the purposes of identifying a potential qualifying single source drug using FDA sources as well as relevant publicly available information as CMS deems appropriate. The potential qualifying single source drug would also include all dosage forms and strengths of the biological product with the same active ingredient and marketed pursuant to the same BLA(s) described in the prior sentences that are: (1) repackaged and relabeled products that are marketed pursuant to such BLA(s); (2) authorized generic drugs, the definition of which at section 1192(e)(2)(B)(ii) of the Act and proposed § 429.20 (described further later in this section) includes unbranded biological products that are marketed pursuant to such BLA(s); or (3) MMA products imported under section 801(d)(1)(B) of the FD&C Act that are marketed pursuant to such BLA(s). Any dosage forms and strengths of the biological product with the same active ingredient that are distributed by a private label distributor and marketed pursuant to such BLAs would also be aggregated in the potential qualifying single source drug of that BLA holder consistent with the policies for implementation as described in, for example, section 30.1 of the Negotiation Program Guidance.
Although assessing biological products on the basis of their active ingredient is appropriate in most circumstances, we understand that a discrete category of biological products—namely, vaccines for infectious disease(s)—are more appropriately assessed using their antigen component due to the evolving nature of pathogen strains over time. Therefore, and as proposed at § 429.125(b)(3), in the context of vaccines for infectious disease(s), we would identify a potential qualifying single source drug on the basis of such vaccines' antigen component on such vaccines' labeling, as accessed in public sources such as those discussed later in this section. We believe our proposal to identify drugs based on their active moiety and biological products based on their active ingredient, subject to the proposal for vaccines for infectious disease(s), is the best reading of the statutory directives in sections 1192(d)(3)(B), 1192(e), 1194(e)(1)(D), and 1196(a)(2) of the Act as they relate to identifying qualifying single source drugs. We note further that in the context of drugs, “active moiety” (as contrasted with “active ingredient”) describes the active molecule or ion in the drug, as this term excludes those appended portions of the molecule that cause the drug to be an ester, salt (including a salt with hydrogen or coordination bonds), or other noncovalent derivative (such as a complex, chelate, or clathrate) of the molecule.[27]
The term “active moiety” is not applicable in the context of biological products, and it is thus appropriate to evaluate biological products based on their active ingredient.
Consistent with the policies for implementation as described in Negotiation Program Guidance, we are proposing to use public sources such as, but not limited to, RxNorm, OpenFDA, FDALabel, DailyMed, and FDA's Active Ingredient-Active Moiety Relationship/Basis of Strength file to identify the active ingredient/active moiety/antigen component of the drug or biological product. We may also consult with FDA as appropriate, for example, to clarify whether a suffix or prefix in an active moiety/active ingredient/antigen component name represents a genuine difference in active moiety/active ingredient/antigen component.
Section 1192(e)(2)(A) of the Act states that an authorized generic drug and the qualifying single source drug that includes the listed drug or reference product of that authorized generic drug shall be treated as the same qualifying single source drug. An authorized generic drug is defined in section 1192(e)(2)(B) of the Act and in proposed § 429.20 as: (1) in the case of a drug, an authorized generic drug (as such term is defined in section 505(t)(3) of the FD&C Act); and (2) in the case of a biological product, a product that has been licensed under section 351(a) of the PHS Act [28]
and is marketed, sold, or distributed directly or indirectly to the retail class of trade under a different labeling, packaging (other than repackaging as the reference product in blister packs, unit doses, or similar packaging for institutions), product code, labeler code, trade name, or trademark than the reference product. We are proposing at § 429.125(b)(1)(iii) that a potential qualifying single source drug that is a drug is inclusive of authorized generic drugs that are marketed under the NDA(s) described therein, and at proposed § 429.125(b)(2)(iii) that a potential qualifying single source drug that is a biological product is inclusive of authorized generic drugs that are marketed under the BLA(s) described therein.
(1) Fixed Combination Drugs
At proposed § 429.125(b)(4), for the purpose of identifying potential qualifying single source drugs and subject to the narrow modification for certain fixed combination drugs proposed at § 429.125(b)(4)(i) to clarify our treatment of new formulations, we
( printed page 36258)
propose that if a drug is a fixed combination drug (defined in proposed § 429.20) with two or more active moieties, active ingredients, or, for vaccines for infectious disease(s), antigen components, the distinct combination of active moieties, active ingredients, or antigen components would generally be treated as one active moiety, active ingredient, or antigen component. Therefore, all formulations with this distinct combination offered by the same NDA/BLA holder would be aggregated across all dosage forms and strengths of the fixed combination drug (that is, all formulations and dosage forms and strengths of this distinct combination would be considered the same potential qualifying single source drug and, if applicable, CMS would aggregate total expenditures for all such formulations and dosage forms and strengths when determining whether such drug is a negotiation-eligible drug or selected drug). Under this proposal, a product containing only one (but not all) of the active moieties, active ingredients, or antigen components that is offered by the same NDA/BLA holder would generally not be aggregated with the formulations of the fixed combination drug and would be considered a separate potential qualifying single source drug. For example, a corticosteroid inhaler would not be aggregated with a fixed combination inhaler from the same NDA/BLA holder that contains the same corticosteroid combined with a long-acting beta agonist. In this example, the corticosteroid inhaler would be considered as a separate potential qualifying single source drug from the fixed combination inhaler.
In the draft guidance for initial price applicability year 2028 and manufacturer effectuation of the MFP in 2026, 2027, and 2028, we stated our belief that treating distinct combinations of active moieties/active ingredients as one active moiety/active ingredient for the purpose of identifying potential qualifying single source drugs is generally appropriate. However, we acknowledged that there may exist fixed combination drugs for which one of the active ingredients or active moieties contained is not biologically active against the disease state(s) the drug is indicated for and thus does not result in a clinically meaningful difference. We solicited comments on whether the addition of drugs payable under Part B may impact the fixed combination drug policy described in the draft guidance. In particular, we solicited comments on how CMS might consider grouping such fixed combination drug products with products containing at least one but not all of the active moiety(ies)/active ingredient(s) into the same potential qualifying single source drug for both drugs payable under Part B and/or covered under Part D, including input on terminology that could facilitate the effectuation of such a policy.
We received many comments in response to this comment solicitation. Some commenters supported a modification to the fixed combination drug policy as a way to close a loophole for manufacturers to avoid selection by making minor changes to existing drugs, specifically citing the addition of hyaluronidase to drugs payable under Part B. Other commenters opposed such a modification, citing concerns that CMS lacks the statutory authority to do so, that the approach described in the comment solicitation does not align with how FDA regulates and reviews the approvals of fixed combination drugs, or that CMS would not be able to apply a consistent definition to the terminology described in comment solicitation, including the terms “biologically active against the disease state” or “clinically meaningful difference”. Some commenters raised concerns that the modification would potentially harm patients and discourage the innovation of pharmaceutical manufacturers. In the Negotiation Program Guidance, we stated that, due to the complexity and scope of the issue as noted in the stakeholder comments, we believed additional time would be necessary to develop objective policy criteria if we were to finalize such a policy, and thus did not make a change to the fixed combination drug policy in the Negotiation Program Guidance. We stated our intent to address the program integrity risk posed by certain fixed combination drugs and that we were continuing to consider appropriate policy to potentially propose in rulemaking for initial price applicability year 2029 and subsequent years.
Since publishing the Negotiation Program Guidance, we have continued to consider program integrity risks posed by certain fixed combination drugs. Specifically, we are aware of a program integrity risk in which, under Negotiation Program policies for identifying qualifying single source drugs set forth in guidance with respect to previous initial price applicability years, a manufacturer may avoid selection of a qualifying single source drug or, in the case that a qualifying single source drug becomes a selected drug, avoid the application of the MFP, by marketing a new formulation of the qualifying single source drug that includes, in addition to the active moiety(ies)/active ingredient(s)/antigen component(s) in the original qualifying single source drug, an active moiety, antigen component, or active ingredient, such as hyaluronidase, that enables an alternative route of administration for the shared active moiety(ies)/active ingredient(s)/antigen component(s) in the original qualifying single source drug. Under the general fixed combination drug policy set forth in guidance with respect to previous initial price applicability years, including, for example, section 30.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028, CMS is aware that such a manufacturer may avoid having all dosage forms and strengths of a drug, including the new formulation of such drug, with its active moiety(ies)/active ingredient(s)/antigen component(s) included in a qualifying single source drug in instances in which a new formulation differs from other formulations of the qualifying single source drug based on the inclusion of an additional active moiety/active ingredient/antigen component that enables an alternative route of administration for the shared active moiety(ies)/active ingredient(s)/antigen component(s).
In this scenario, we are concerned that application of CMS' general fixed combination drug policy would be in tension with statutory requirements under sections 1192(d)(3)(b) and 1196(a)(2) of the Act because we would not aggregate together the original qualifying single source drug and the new formulation containing the additional active moiety/active ingredient/antigen component, even though these may be appropriately understood as different formulations of the same drug under sections 1192(d)(3)(B) and 1196(a)(2) of the Act.
First, application of the general fixed combination drug policy to such products would be in tension with section 1192(d)(3)(B) of the Act in the course of drug selection because we would not treat the original qualifying single source drug and the new formulation containing the additional active moiety/active ingredient/antigen component as one qualifying single source drug. Thus, when determining whether a qualifying single source drug is a negotiation-eligible drug, as proposed at § 429.115, we would calculate the combined total expenditures under Part B and Part D separately for the original qualifying single source drug and the new formulation. We believe that separately calculating total expenditures for such products under these circumstances
( printed page 36259)
may be inconsistent with the statutory direction at section 1192(d)(3)(B) of the Act to use data that is aggregated across dosage forms and strengths of the drug, including new formulations of the drug, in determining whether a qualifying single source drug is a negotiation-eligible drug. Because under the general fixed combination drug policy we would evaluate the original qualifying single source drug and the new formulation separately during the drug selection process, the time since approval or licensure (determined as proposed at § 429.125(c)) for the new formulation would likely be later than for the original qualifying single source drug, thereby extending the years before which the new formulation could potentially be selected and, if selected and if an MFP is agreed to, be subject to an MFP. This dynamic presents program integrity risks because evaluating the time since approval or licensure separately for the original qualifying single source drug and the new formulation and splitting combined total expenditures under Part B and Part D between such products could lead to a scenario where the original qualifying single source drug or the new formulation or both are never selected or are not selected until a future initial price applicability year, which would reduce or delay the ability of the Negotiation Program to negotiate MFPs for high expenditure, single source drugs and biological products.
Second, if the original qualifying single source drug is selected for negotiation, and a negotiated MFP is agreed upon for that drug, the general fixed combination drug policy may be in tension with section 1196(a)(2) of the Act, which directs CMS to apply the MFP across different strengths and dosage forms of a selected drug and not based on the specific formulations or package size or package type of such drug, because the MFP would not apply to the new formulation. This dynamic presents further program integrity risks. If the new formulation is not part of the selected drug, then the Primary Manufacturer would be able to market the new formulation without the new formulation being subject to MFP effectuation requirements, thereby creating a new incentive for the Primary Manufacturer to drive patient utilization toward the new formulation and circumvent the application of the MFP to the original qualifying single source drug for such patients.
To address these program integrity risks, and to clarify how CMS interprets sections 1192(d)(3)(B) and 1196(a)(2) of the Act as applied to new formulations of a drug, including in the context of fixed combination drugs, we are proposing at § 429.125(b)(4)(i) a narrow modification to the application of the general fixed combination drug policy: if CMS determines that a fixed combination drug with two or more active moiety(ies), active ingredient(s), or, for a vaccine for infectious disease(s), antigen component(s) shares one or more active moiety(ies), active ingredient(s), or antigen component(s) with another drug or biological product(s) with the same NDA/BLA holder, and such products differ in active moiety(ies), active ingredient(s), or antigen component(s) due to the inclusion of an active moiety, active ingredient, or antigen component that creates a new formulation and enables an alternative route of administration for the co-administered active moiety(ies), active ingredient(s), or antigen component(s), CMS would, for purposes of identifying the potential qualifying single source drug under proposed § 429.125(b)(1) and (b)(2), use all dosage forms and strengths of the drug or biological product with the shared moiety(ies), active ingredient(s), or antigen component(s) and the same NDA/BLA holder. In other words, we would identify the potential qualifying single source drug as including all dosage forms and strengths with the shared active moiety(ies), active ingredient(s), or antigen component(s) that is offered by the same NDA/BLA holder. An example is the inclusion of active ingredient X with a different active ingredient Y, where active ingredient X creates a new formulation and enables an alternative route of administration for active ingredient Y. In the described example and as set forth in proposed § 429.125(b)(4)(i), CMS would identify the potential qualifying single source drug using all dosage forms and strengths of the drug or biological product that contain active ingredient Y and share the same BLA holder.[29]
We are proposing the policy at § 429.125(b)(4)(i) at this time because the program integrity concerns this policy would address relate primarily to biologics that are more likely payable under Part B, and drugs were not selected based on total expenditures under Part B until initial price applicability year 2028. The program integrity concerns raised by fixed combination drugs that are new formulations have not yet impacted drug selection but may soon as more drugs with significant total expenditures under Part B become qualifying single source drugs and negotiation-eligible drugs. We do not believe that significant reliance interests have arisen around the fixed combination policy established in Negotiation Program guidance for initial price applicability years 2026 through 2028, as incentives for pharmaceutical manufacturers to develop new formulations like the ones we propose to address in this policy long predate the Negotiation Program. In the absence of this proposed policy, however, pharmaceutical manufacturers may have increased incentives under Negotiation Program policy to drive patient utilization toward new formulations that would be treated as distinct qualifying single source drugs, and if selected, as distinct selected drugs.
This proposal incorporates feedback from commenters that raised concerns about how we would operationalize terms used in the draft guidance for initial price applicability year 2028 and manufacturer effectuation of the MFP in 2026, 2027, and 2028, such as “biologically active against the disease state”. We understand from these commenters' concerns the importance of developing a modification to the general fixed combination drug policy that is narrowly tailored to the development of new formulations that pose program integrity concerns and does not group products together under the same potential qualifying single source drug beyond what the statute permits. Additionally, we understand the importance of developing criteria that allow the narrow modification to be operationalized in a consistent and objective manner over a large set of active moieties/active ingredients/antigen components. We believe that the terminology proposed at § 429.125(b)(4)(i) would achieve both these goals.
First, we believe the criteria proposed at § 429.125(b)(4)(i) can be operationalized in a consistent and objective manner using public sources. Specifically, we are proposing to use public sources such as, but not limited to, RxNorm, OpenFDA, FDALabel, DailyMed, and FDA's Active Ingredient-Active Moiety Relationship/Basis of Strength file to identify the active moiety(ies)/active ingredient(s)/antigen component(s) of the drug or biological products, as described in section II.B.6.a. of this proposed rule, and to use public sources such as, but not limited
( printed page 36260)
to, Orange Book, Purple Book, Drugs@FDA, and DailyMed to identify the routes of administration of the fixed combination drug and the drug or biological product with the same NDA/BLA holder that shares one or more active moiety(ies)/active ingredient(s)/antigen component(s) with the fixed combination drug. We may also consult with FDA, as appropriate. We solicit comments on recommended approaches to inform CMS identification of active moiety(ies)/active ingredient(s)/antigen component(s) that enable a new route of administration for co-administered active moiety(ies)/active ingredient(s)/antigen component(s), including what types of information or descriptions we should look for within product labeling when identifying such cases.
Second, focusing on active moieties/active ingredients/antigen components that enable an alternative route of administration would reduce the number of products qualifying for the narrow modification as compared to the previous language citing active moieties/active ingredients not biologically active against the disease state(s) for which the drug is indicated. For example, we believe that the terminology proposed at § 429.125(b)(4)(i) would currently only impact biological products licensed in BLAs.
In developing this proposed policy, CMS also considered other feedback from commenters on our prior proposal to modify our fixed combination drug policy. As we noted in the Negotiation Program Guidance, many commenters raised concerns that a modification to CMS' fixed combination drug policy would not align with how FDA regulates and reviews the approvals of fixed combination drugs. We considered this feedback but maintain that the language at proposed § 429.125(b)(4)(i) clarifies how CMS would comply with sections 1192(d)(3)(B) and 1196(a)(2) of the Act for fixed combination drugs that are new formulations, and the commenters' suggestion that CMS must treat fixed combination drugs in the same manner as what such commenters assert is how the FDA may treat such products under FDA authorities is not persuasive in this instance. This proposal to identify such products as a single potential qualifying single source drug reflects CMS' proposed interpretation of sections 1192(d)(3)(B) and 1196(a)(2) of the Act and would solely be for the purposes of implementing the Negotiation Program.
We also considered comments on how a modification to CMS' fixed combination drug policy would potentially impact pharmaceutical innovation. Although a few commenters stated a modification to the fixed combination drug policy would potentially harm patients by discouraging the innovation of pharmaceutical manufacturers, another commenter noted that manufacturers already have substantial financial motivations beyond the Negotiation Program to pursue the development of new formulations—including new formulations that are fixed combination drugs—to delay generic or biosimilar competition. We agree with this latter argument and note further our belief that the policy proposed at § 429.125(b)(4)(i) would effectuate section 1192(d)(3)(B) and 1196(a)(2) of the Act, address the program integrity concerns in the Negotiation Program that we and commenters have identified, while not meaningfully impacting incentives for innovation. We also reiterate our statement in the Negotiation Program Guidance that we are committed to a negotiation process that recognizes the clinical benefit of products, including products with different dosage forms and strengths, formulations or routes of administration from other products that are aggregated as part of the same qualifying single source drug, and we direct readers to proposed § 429.510(e) and section II.F.3.d.1. of this proposed rule for discussion of CMS' approach to adjusting the starting point for an initial offer based on section 1194(e)(2) factors, which includes factors related to clinical benefit as compared to therapeutic alternatives.
(2) Alternatives Considered
We considered expressly limiting the proposed modification of the application of the general fixed combination drug policy to biological products (other than vaccines for infectious disease(s) identified at proposed § 429.125(b)(3)), as currently such biological products licensed in BLAs pose the program integrity risks of which we are currently aware. However, in line with our aim to apply the statutory requirements at sections 1192(d)(3)(B) and 1196(a)(2) of the Act equally to all new formulations of a drug, whether the new formulation is approved in an NDA or licensed in a BLA, and to account for the possibility that the narrow modification for fixed combination drugs proposed at § 429.125(b)(4)(i) could apply to drugs in the future, we are not proposing to limit the proposed modification to these biological products. This approach would ensure that we are treating new formulations equally, whether that new formulation is a drug or biological product.
We also considered proposing an alternative modification to the general fixed combination drug policy that would target the program integrity risk as described in the Negotiation Program Guidance wherein CMS identification of potential qualifying single source drugs under the fixed combination drug policy would not take into account an active moiety, active ingredient, or antigen component that is not biologically active against the disease state(s) the drug is indicated for and thus does not result in a clinically meaningful difference from other drug or biological products that otherwise have the same active moiety(ies)/active ingredient(s)/antigen component(s). However, we are instead proposing to focus on products that differ in active moiety(ies)/active ingredient(s)/antigen component(s) due to the inclusion of an active moiety/active ingredient/antigen component that creates a new formulation and enables an alternative route of administration for the co-administered active moiety(ies)/active ingredient(s)/antigen component(s). As noted in section II.B.6.a.1. of this proposed rule, we believe the proposed approach narrowly addresses the program integrity risks with the general fixed combination policy and the specific elements of the policy that are in tension with section 1192(d)(3)(B) of the Act, which directs CMS to aggregate across dosage forms and strengths, including new formulations of the drug, and section 1196(a)(2) of the Act, which directs CMS to apply the MFP across different strengths and dosage forms of a selected drug and not based on the specific formulations or package size or package type of such drug.
We considered maintaining the policy established for initial price applicability years 2026 through 2028, wherein, for a fixed combination drug with two or more active moieties/active ingredients/antigen components, we would treat the distinct combination of active moieties/active ingredients/antigen components as one active moiety/active ingredient/antigen component. However, for the reasons described in section II.B.6.a.1. of this proposed rule, we believe that this policy poses program integrity risks and is in tension with sections 1192(d)(3)(B) and 1196(a)(2) of the Act.
Finally, we considered proposing policy to address program integrity risks posed by co-packaged drugs. In the Negotiation Program Guidance, we stated that the general fixed combination drug policy would apply to a co-packaged drug, in which two active moieties/active ingredients are not co-
( printed page 36261)
formulated but rather co-packaged and sold in a single package, but that we may address co-packaged drugs in rulemaking for initial price applicability year 2029 and subsequent years. That is, for purposes of the Negotiation Program in initial price applicability year 2028, if a drug (including a co-packaged drug) contained two or more active moieties/active ingredients/antigen components, the distinct combination of active moieties/active ingredients/antigen components would be considered as one active moiety/active ingredient/antigen components for the purpose of identifying potential qualifying single source drugs, whether such drug was co-formulated or co-packaged. After further consideration, and based on our assessment that at this time co-packaged drugs do not appear to pose substantive program integrity risks to the Negotiation Program because manufacturers are not adopting co-packaging practices that abuse current program policies, we are proposing that the general fixed combination drug policy at proposed § 429.125(b)(4) continue to apply to co-packaged drugs. We are monitoring this approach and potential gaming of co-packaged drugs and may consider revisiting this policy in the future.
We solicit comments on our proposal and these alternatives.
b. Time Since Approval or Licensure (§ 429.125(c))
In accordance with section 1192(e)(1) of the Act and consistent with policies for implementation as described in Negotiation Program Guidance, and accounting for the treatment of certain former orphan drugs in accordance with section 1192(e)(4) of the Act as addressed in proposed § 429.125(c)(3), we are proposing at § 429.125(c)(1) and (c)(2) that at least 7 years (for drugs) or 11 years (for biological products) must have elapsed between the FDA date of approval or licensure of the potential qualifying single source drug identified at proposed § 429.125(b) and the selected drug publication date with respect to an initial price applicability year. To determine the date of approval for a potential qualifying single source drug that is a drug with more than one FDA application number, we would use the initial date of approval associated with the earliest-approved FDA application belonging to the NDA holder and containing the active moiety (or in the case of a potential qualifying single source drug identified under the general fixed combination drug policy proposed at § 429.125(b)(4), for the distinct combination of active moieties). For a potential qualifying single source drug identified under § 429.125(b)(4)(i) that is a drug, we would use the initial date of approval associated with the earliest-approved FDA application belonging to the NDA holder and containing the shared active moiety(ies) used to identify the potential qualifying single source drug. To determine the date of licensure for a potential qualifying single source drug that is a biological product (except in the case of a vaccine for infectious disease(s), for which we would determine the date of licensure as described later in this section) with more than one FDA application number, we would use the initial date of licensure associated with the earliest-approved FDA application belonging to the BLA holder and containing the active ingredient (or in the case of a potential qualifying single source drug identified under the general fixed combination drug policy proposed at § 429.125(b)(4), for the distinct combination of active ingredients). For a potential qualifying single source drug identified under § 429.125(b)(4)(i) that is a biological product, we would use the initial date of licensure associated with the earliest-approved licensure of the FDA application belonging to the BLA holder and containing the shared active ingredient(s) used to identify the potential qualifying single source drug.[30]
Consistent with prior initial price applicability years, we are also proposing that the identification of the earliest-approved FDA application belonging to the NDA/BLA holder and containing the active moiety/active ingredient, respectively, would be made irrespective of whether the indication(s) approved in such NDA/BLA were or are covered under Part D or payable under Part B or both. We would determine whether a product is not covered under Part D or payable under Part B at the NDC-11 level via the Primary Manufacturer's submission of information on NDC-11s as proposed at § 429.100(d) and described in section II.B.1. of this proposed rule.
As described in section II.B.6.a. of this proposed rule, to give full effect to all relevant provisions of the statute, including sections 1192(d)(3)(B), 1192(e), 1194(e)(1)(D), and 1196(a)(2) of the Act, we are proposing to identify a potential qualifying single source drug using the specific constituent dosage forms and strengths that are identified as aggregated under the same NDA/BLA holder for the same active moiety/active ingredient, inclusive of products that are marketed under different NDAs/BLAs. Therefore, to identify the date of approval or licensure, as applicable, for a potential qualifying single source drug with more than one FDA application, sections 1192(e)(1)(A)(ii) and 1192(e)(1)(B)(ii) of the Act require CMS to use the initial approval or licensure date associated with the earliest-approved FDA application for such potential qualifying single source drug to ensure that CMS captures the full time since the earliest product in the potential qualifying single source drug was approved or licensed.
To provide an example for how we would determine the approval or licensure date set forth at proposed § 429.125(c)(1) and (c)(2), respectively, the selected drug publication date for initial price applicability year 2029 is February 1, 2027, consistent with section 1191(b)(3) of the Act and proposed § 429.20. As such, for initial price applicability year 2029, the initial date of approval for a drug to be considered a qualifying single source drug must have been on or before February 1, 2020, and the initial date of licensure for a biological product to be considered a qualifying single source drug must have been on or before February 1, 2016.
We are aware that some manufacturers of vaccines for infectious disease(s) update the antigen component(s) of their products through supplemental BLAs. Consistent with the policies for implementation as described in Negotiation Program Guidance, for each unique potential qualifying single source drug that CMS identifies based on its antigen component(s) in accordance with proposed § 429.125(b)(3), we are proposing at § 429.125(c)(2)(i) that CMS will use the initial date of licensure for any BLA or supplemental BLA for that unique potential qualifying single source drug for purposes of identifying the starting date from which to measure the 11 years described in proposed § 429.125(c)(2).
Additionally, consistent with the policies for implementation as described in Negotiation Program Guidance and as proposed in § 429.125(c)(2)(ii), for biological products with an approved NDA that was deemed to be a BLA on March 23, 2020, in accordance with section 7002(e)(4)(A) of Biologics Price Competition and Innovation Act of 2009 (BPCI Act), and that are currently licensed biological products under
( printed page 36262)
section 351 of the PHS Act (hereinafter “deemed biologics”), we would consider March 23, 2020 to be the licensure date for purposes of identifying the starting date from which to measure the 11 years described in proposed § 429.125(c)(2).[31]
In accordance with section 1192(e)(4) of the Act and as proposed at § 429.125(c)(3), if, as of the date of a drug or biological product's initial approval or licensure, such drug or biological product met or meets the criteria for the orphan drug exclusion proposed at § 429.125(e)(1), we would measure the 7- and 11-year periods identified in proposed § 429.125(c)(1) and (c)(2) starting from the first day after such initial date of approval or licensure that such drug or biological product does not, or did not, meet the criteria for the orphan drug exclusion at proposed § 429.125(e)(1). We would identify this day as the earlier of (1) the date on which the FDA approves such drug or biological product for an indication for a disease or condition that is not a rare disease or condition for which the drug or biological product is designated under section 526 of the FD&C Act; or (2) the date on which an orphan drug designation is withdrawn, if that withdrawal results in the drug or biological product no longer qualifying for the orphan drug exclusion.[32]
As noted previously, section 1192(e)(4) of the Act requires that CMS measure the 7- and 11-year periods set forth in proposed § 429.125(a)(1)(ii) and (a)(2)(ii) starting from “the first day” after the drug's initial date of approval or licensure that such drug or biological product does not, or did not, meet the criteria for the orphan drug exclusion. We are thus proposing that the determination of the date on which the FDA approves the drug or biological product for an indication for a disease or condition that is not a rare disease or condition for which the drug or biological product is designated under section 526 of the FD&C Act would be made irrespective of whether approval of such indication is later withdrawn. We are aware that this proposal could result in a scenario where the 7- and 11-year periods set forth in proposed § 429.125(a)(1)(ii) and (a)(2)(ii) would not yet have elapsed between the last day that the drug most recently qualified for the orphan drug exclusion at section 1192(e)(3)(A) of the Act and proposed at § 429.125(e)(1) and the selected drug publication date, but we believe this approach aligns best with statute given the requirement at section 1192(e)(4) of the Act to measure the 7- and 11-year periods starting from “the first day” after the drug's initial date of approval or licensure that such drug or biological product does not, or did not, meet the criteria for the orphan drug exclusion. We are not currently aware of any instances in which this scenario—wherein the 7- and 11-year periods would not yet have elapsed between the last day that the drug most recently qualified for the orphan drug exclusion and the selected drug publication date—would occur in practice and believe the chances of this scenario occurring in the future are low.
c. Exclusions From Qualifying Single Source Drugs (§ 429.125(e))
Section 1192(e)(3) of the Act requires that the term “qualifying single source drug” not include certain orphan drugs described in section 1192(e)(3)(A) of the Act, low-spend Medicare drugs described in section 1192(e)(3)(B) of the Act, or plasma-derived products described in section 1192(e)(3)(C) of the Act. With respect to initial price applicability years 2026 through 2028, CMS implemented these requirements through guidance, including, for example, sections 30.1.1 through 30.1.3 of the Negotiation Program Guidance with respect to initial price applicability year 2028. We are proposing at § 429.125(e) to exclude certain drugs when identifying qualifying single source drugs.
(1) Orphan Drug Exclusion From Qualifying Single Source Drugs (§ 429.125(e)(1))
Section 1192(e)(3)(A) of the Act excludes certain orphan drugs from the definition of a qualifying single source drug (hereinafter “the orphan drug exclusion”). As such, and in accordance with section 30.1.1 of the Negotiation Program Guidance, in proposed § 429.125(e)(1), we propose to codify the existing policy established in section 30.1.1 of the Negotiation Program Guidance, which incorporated amendments to the orphan drug exclusion in accordance with the “Working Families Tax Cut” legislation (Pub. L. 119-21), to exclude from the definition of a qualifying single source drug a drug or biological product that is designated as a drug for one or more rare diseases or conditions under section 526 of the FD&C Act and for which the only approved indication (or indications) [33]
is for one or more such rare diseases or conditions (as such term is defined in section 526(a)(2) of the FD&C Act and proposed § 429.20).
To be considered for the orphan drug exclusion with respect to each initial price applicability year beginning with initial price applicability year 2029, the drug or biological product must: (1) be designated as a drug for one or more rare diseases or conditions under section 526 of the FD&C Act; and (2) be approved by the FDA only for one or more indications within such designated rare disease(s) or condition(s). We would not consider withdrawn orphan drug designations or withdrawn approvals when determining whether a drug meets the orphan drug exclusion.[34]
To determine whether a potential qualifying single source drug qualifies for the orphan drug exclusion, we propose to consider all dosage forms and strengths of the potential qualifying single source drug, as described previously and as identified in proposed § 429.125(b). We would use the FDA Orphan Drug Product designation database [35]
and information on FDA-approved indications from other publicly available databases and documents (such as, but not limited to, FDALabel,[36]
FDA Online Label Repository,[37]
Drugs@FDA,[38]
and NLM DailyMed [39]
) to determine whether a drug meets the requirements in section 1192(e)(3)(A) of the Act to qualify for the orphan drug exclusion. We would also consult with FDA, as appropriate, including as to whether the approved indication(s) is for a rare disease(s) or
( printed page 36263)
condition(s) for which the drug or biological product is designated under section 526 of the FD&C Act. We would not consider whether an FDA-approved indication was or is covered under Part D or payable under Part B or both when determining whether a drug meets the requirements in section 1192(e)(3)(A) of the Act to qualify for the orphan drug exclusion.
(2) Low-Spend Medicare Drug Exclusion From Qualifying Single Source Drugs (§ 429.125(e)(2))
Section 1192(e)(3)(B) of the Act also excludes low-spend Medicare drugs or biological products with combined expenditures under Medicare Part B and Part D during the total expenditures measurement period less than the inflation-adjusted threshold for the previous initial price applicability year, increased by the annual percentage increase in the CPI-U for the 12-month period ending on September 30 of the year prior to the year of the selected drug publication date with respect to a given initial price applicability year, when identifying qualifying single source drugs (“the low-spend Medicare drug exclusion”). As such, in proposed § 429.125(e)(2), we propose to codify the policy established in section 30.1.2 of the Negotiation Program Guidance to exclude such low-spend Medicare drugs or biological products from the definition of a qualifying single source drug.
To identify drugs and biological products meeting the statutory criteria for the low-spend Medicare drug exclusion with respect to each initial price applicability year beginning with initial price applicability year 2029, we propose to calculate combined total expenditures under Part B and Part D for a potential qualifying single source drug as the sum of total expenditures under Part B plus total expenditures under Part D, calculated as set forth in proposed § 429.120 and described in section II.B.5. of this proposed rule. We would exclude from the final list of qualifying single source drugs for the initial price applicability year any drugs for which the sum of total expenditures under Part B and Part D is less than the inflation-adjusted threshold for that initial price applicability year. As set forth in sections 1192(e)(3)(B)(i) and (e)(3)(B)(ii) of the Act and codified in proposed § 429.125(e)(2)(ii), the inflation-adjusted threshold for an initial price applicability year is equal to the inflation-adjusted threshold for the previous initial price applicability year, increased by the annual percentage increase in the CPI-U for the 12-month period ending on September 30 of the year prior to the year of the selected drug publication date for such initial price applicability year, starting from the inflation-adjusted threshold for initial price applicability year 2028 equal to $212,907,518.30.
(3) Plasma-Derived Product Exclusion From Qualifying Single Source Drugs (§ 429.125(e)(3))
Additionally, section 1192(e)(3)(C) of the Act excludes plasma-derived products from the definition of a qualifying single source drug. As such, in proposed § 429.125(e)(3), we propose to codify the existing policy established in section 30.1.3 of the Negotiation Program Guidance to exclude plasma-derived products when identifying qualifying single source drugs (“the plasma-derived product exclusion”) with respect to each initial price applicability year beginning with initial price applicability year 2029. Under section 1192(e)(3)(C) of the Act, a plasma-derived product is a biological product that is derived from human whole blood or plasma. In implementing this statutory exclusion, we would identify plasma-derived products by referring to product information, including approved product labeling, available on the FDA Approved Blood Products website, including the list of fractionated plasma products,[40]
and would refer to databases such as, but not limited to, FDALabel [41]
and the FDA Online Label Repository [42]
to verify if the product is derived from human whole blood or plasma. CMS also would consult with FDA, as appropriate. Consistent with existing policy under Negotiation Program Guidance, for purposes of applying the exclusion, we consider only whether the active moiety/active ingredient is derived from human whole blood or plasma.
d. Bona Fide Marketing of an Approved Generic Drug or Licensed Biosimilar and Deselection of a Selected Drug (§ 429.125(d), § 429.130, and § 429.135)
Key provisions of the Negotiation Program statute govern CMS' evaluation of generic and biosimilar competitors to potential qualifying single source drugs and CMS' further consideration of such products in the deselection of selected drugs. First, section 1192(e)(1)(A)(iii) of the Act states that, to be considered a qualifying single source drug, a drug cannot be the listed drug for any drug approved and marketed under an ANDA under section 505(j) of the FD&C Act. For a biological product, section 1192(e)(1)(B)(iii) of the Act states that the biological product cannot be the reference product for any biological product that is licensed and marketed under section 351(k) of the PHS Act (that is, a biosimilar). Second, section 1192(c)(1) specifies a selected drug that is included on the list of selected drugs for an initial price applicability year will remain a selected drug for that year and each subsequent year beginning before the first year that begins at least nine months after the date on which CMS determines: (1) the FDA has approved a generic drug under section 505(j) of the FD&C Act that identifies as its reference-listed drug a product that is included in the selected drug, or FDA has licensed a biosimilar under section 351(k) of the PHS Act that identifies as its reference product a product that is included in the selected drug; and (2) the generic drug or biosimilar, as applicable, is marketed pursuant to such approval or licensure.
Based on the authorities described previously, CMS conducts the following inquiry to determine whether a drug or biological product can be considered a qualifying single source drug. First, using FDA reference sources including the Orange Book and Purple Book, CMS would evaluate whether at least one generic drug is approved under section 505(j) of the FD&C Act using any dosage form or strength of the potential qualifying single source drug as the listed drug or at least one biosimilar is licensed under section 351(k) of the PHS Act using any dosage form or strength of the potential qualifying single source drug as the reference product, consistent with proposed § 429.125(d)(1). This approach is consistent with the approach of aggregating different dosage forms and strengths as a single potential qualifying single source drug as required under section 1192(d)(3)(B) of the Act (stating that a qualifying single source drug is inclusive of all strengths and dosage forms, which is described in further detail in section II.B.6.a. of this proposed rule). In other words, consistent with proposed in § 429.125(d), if CMS determines there is a generic drug or biosimilar that is approved or licensed, as applicable, for any dosage form or strength of a drug or biological product, and such generic drug or biosimilar is subject to Bona Fide Marketing, then that drug or biological product would not meet the statutory criteria in section 1192(e)(1)(A)(iii) or section 1192(e)(1)(B)(iii) of the Act to be a
( printed page 36264)
qualifying single source drug. We are proposing that the determination of whether a potential qualifying single source drug includes the listed drug or reference product for an approved generic drug or licensed biosimilar, respectively, would be made irrespective of whether the indication(s) approved for the listed drug or reference product were or are covered under Part D, payable under Part B, or both.
We note that because section 1192(e)(2)(A) of the Act provides that an “authorized generic” drug “shall be treated as the same qualifying single source drug,” we will not consider an authorized generic drug to be a generic drug or biosimilar for the purposes of identifying whether there is an approved generic drug or licensed biosimilar for any strength or dosage form of a potential qualifying single source drug. An authorized generic drug is defined in section 1192(e)(2)(B) of the Act as: (1) in the case of a drug, an authorized generic drug (as such term is defined in section 505(t)(3) of the Federal Food, Drug, and Cosmetic Act); and (2) in the case of a biological product, a product that has been licensed under section 351(a) of such Act [43]
and is marketed, sold, or distributed directly or indirectly to retail class of trade under a different labeling, packaging (other than repackaging as the reference product in blister packs, unit doses, or similar packaging for use in institutions), product code, labeler code, trade name, or trade mark than the reference product.
Second, consistent with the policies for implementation of section 30.1 of the Negotiation Program Guidance, we would conduct a holistic inquiry based on the totality of the circumstances when evaluating whether the manufacturer(s) of any approved generic drug(s) or licensed biosimilar(s) is or are engaged in Bona Fide Marketing of that generic drug or biosimilar as defined at proposed § 429.20 and described in proposed § 429.130. CMS must evaluate, as part of the determination described in section 1192(e) of the Act, whether a generic drug or biosimilar “is . . . marketed” and whether the potential qualifying single source drug is a listed drug “for any drug that is approved and marketed under section 505(j) of the FD&C Act” or a reference product “for any biological product that is licensed and marketed under section 351(k) of the PHS Act.” The terminology specified in sections 1192(e) of the Act for purposes of the Negotiation Program is distinct from the terminology specified elsewhere in the Act for purposes of the Medicare Part B Inflation Rebate Program (section 1847A(i) of the Act) and the Medicare Part D Inflation Rebate Program (section 1860D-14B of the Act), also established through the IRA. For Medicare inflation rebates, the statute refers to the date that a drug is “first marketed”. The absence of the term “first marketed” in section 1192 of the Act indicates that, for purposes of the Negotiation Program, a generic drug or biosimilar must have a continuing presence on the market in order to affect the status of a listed drug/reference product. Consistent with the statutory purpose of lowering drug prices through the mechanisms described in Title I, Subtitle B, Part 1 of the Inflation Reduction Act, and as CMS has explained in response to comment on the Negotiation Program Guidance, CMS will consider whether meaningful competition exists on an ongoing basis between any dosage form or strength of a potential qualifying single source drug that includes the listed drug or reference product and any one or more generic drug(s) or biosimilar(s). Therefore, as reflected in proposed § 429.130(a)(1) and (2), we understand that whether a product “is marketed” requires more than solely token or de minimis availability. We appreciate that, while sales and utilization data provide important indicators of market competition, they are not the only sources of information that may illustrate whether such competition exists between a listed drug or reference product and a generic drug or biosimilar. For example, we are also aware there may be situations in which a manufacturer of a brand name drug or biologic has entered into a market-limiting agreement with a manufacturer of a generic drug or biosimilar, where the generic drug or biosimilar manufacturer agrees to limit production or distribution of the generic drug or biosimilar, such that only a nominal quantity of product is allowed to enter the market. The result is a lack of meaningful price competition, and in that circumstance the generic drug or biosimilar is not “marketed” within the meaning of that term as it is used in the section 1192 of the Act (for example, specifically within the context of marketed at section 1192(e) of the Act). Therefore, as proposed in § 429.130(b), whether such competition exists between a listed drug or reference product and an approved generic drug or licensed biosimilar would depend on a holistic inquiry based on the totality of circumstances in existence at the time that CMS evaluates whether an approved generic drug or licensed biosimilar is subject to Bona Fide Marketing and not on the presence, or absence, of any single factor.
Specifically, in accordance with sections 1192(e) of the Act, to consider whether there are ongoing sales and/or utilization of a generic drug or biosimilar in the market, we propose at § 429.130(a)(1)(i) through (iii) to review the following data sources: PDE data, AMP data, and ASP data. Additionally, as part of CMS' holistic consideration of whether one or more manufacturers of a generic drug or biosimilar are engaging in Bona Fide Marketing of the approved generic drug or licensed biosimilar, we propose at § 429.130(a)(1)(iv) that CMS may also review sales and/or utilization data from additional data sources, including, but not limited to, Medicaid State Drug Utilization Data, and data from nationally representative and commercially available databases. Further, to consider whether there is meaningful competition, CMS may also consider other factors proposed at § 429.130(a)(2) and (3) that include whether the approved generic drug or licensed biosimilar is regularly and consistently available for purchase through the pharmaceutical supply chain, whether any licenses or other agreements between a Primary Manufacturer and a generic drug or biosimilar manufacturer limit the availability or distribution of the generic drug or biosimilar, and other available data and informational sources on market share and relative market competition of the approved generic drug or licensed biosimilar that CMS may identify.
If any dosage form or strength of a potential qualifying single source drug is the listed drug or reference product for one or more generic drugs or biosimilars that CMS determines are approved or licensed and subject to Bona Fide Marketing based on the information as described in proposed § 429.130(a), the potential qualifying single source drug would not be considered a qualifying single source drug for the applicable initial price applicability year (as described in proposed § 429.125(d)). As set forth in proposed § 429.125(d) and in accordance with § 429.130(c)(1), we would review the information set forth in proposed § 429.130(a) for any potential qualifying single source drug for initial price applicability year 2029 and any initial price applicability year thereafter in January prior to the selected drug publication date
( printed page 36265)
(proposed at § 429.20 and described in section II.A.2. of this proposed rule). For example, for initial price applicability year 2029, CMS would review this information for any potential qualifying single source drugs in January 2027, prior to the February 1, 2027 selected drug publication date.
Consistent with section 1192(c)(1) of the Act, a drug selected for negotiation for an initial price applicability year would remain a selected drug for that initial price applicability year and each subsequent year beginning before the first year that begins at least nine months after the date as of which CMS determines at least one generic drug is approved under section 505(j) of the FD&C Act using the selected drug as the listed drug or at least one biosimilar is licensed under section 351(k) of the PHS Act using the selected drug as the reference product, and such generic drug or biosimilar is marketed pursuant to such approval or licensure, as applicable. As discussed previously in this section, we understand that whether a product “is marketed” requires more than solely token or de minimis availability. Therefore, when evaluating whether a generic drug or biosimilar for a selected drug is subject to Bona Fide Marketing for the purposes of determining if a selected drug ceases to be a selected drug consistent with section 1192(c) of the Act and proposed § 429.135(a), we would implement the same approach that we propose for evaluating whether a generic drug or biosimilar for a potential qualifying single source drug is subject to Bona Fide Marketing.
If CMS determines that a generic drug is approved or a biosimilar is licensed for the selected drug, and such generic drug or biosimilar is subject to Bona Fide Marketing (as set forth in proposed § 429.130(a)) on a date during the period beginning on the selected drug publication date (as defined in proposed § 429.20) and ending on November 1 of the year that begins 2 years prior to the initial price applicability year for which the drug is selected for negotiation, consistent with section 1192(c)(2) of the Act and proposed § 429.135(b)(1)(i), the selected drug is no longer subject to the negotiation process—including, for example, any applicable negotiation meeting(s) or offer and counteroffer exchanges—for such initial price applicability year. Similarly, consistent with section 1194(f)(5)(B) of the Act and proposed § 429.135(b)(2), if CMS determines that a generic drug is approved or a biosimilar is licensed for a selected drug and that such generic drug or biosimilar is subject to Bona Fide Marketing (as set forth in proposed § 429.130(a)) on a date during the period beginning on the selected drug publication date (as defined in proposed § 429.20) and ending on November 1 of the year that begins 2 years prior to the initial price applicability year for which the drug is selected for renegotiation, then the selected drug is no longer subject to the renegotiation process. Therefore, for drugs selected for negotiation and renegotiation (if any) for an initial price applicability year, we would review the information set forth in proposed § 429.130(a) on a monthly basis starting in March of the calendar year of the negotiation period (or renegotiation period, as applicable) until November 1 of the year that begins 2 years prior to the initial price applicability year for which the drug was selected for negotiation (or renegotiation, as applicable) (see proposed § 429.130(c)(4) and (c)(5)).
Pursuant to section 1192(c)(1) of the Act, once the negotiation period concludes, a selected drug would cease to be a selected drug if CMS determines that a generic drug is approved or a biosimilar is licensed and such generic drug or biosimilar is subject to Bona Fide Marketing based on the information considered in proposed § 429.130(a). We propose that we would review the information set forth in proposed § 429.130(a), biannually in March and October, starting in October of the calendar year after CMS and the Primary Manufacturer reached an agreement on an MFP for the initial price applicability year for which the drug was selected originally for negotiation and until CMS determines that a selected drug meets the requirements at proposed § 429.135(a) to cease being a selected drug.
Section 1194(f)(5) of the Act clarifies that a renegotiation-eligible drug for which CMS determines there is a generic drug that is approved or biosimilar that is licensed, and such generic drug or biosimilar is subject to Bona Fide Marketing before or during the renegotiation period shall not be subject to the renegotiation process. Thus, for any renegotiation-eligible drug, we would review the information set forth in proposed § 429.130(a), in January, prior to the selected drug publication date (defined at § 429.20 and described in section II.A.2. of this proposed rule) as proposed in § 429.130(c)(3).
Finally, after determining that a generic drug is approved or biosimilar is licensed and such generic drug or biosimilar is subject to Bona Fide Marketing, we would monitor that the approved generic drug or licensed biosimilar continues to be subject to Bona Fide Marketing. We would review the information set forth in proposed § 429.130(a) to monitor whether the manufacturer(s) of such generic drug(s) or biosimilar(s) continue to engage in Bona Fide Marketing of such generic drug(s) or biosimilar(s) consistent with the schedule proposed at § 429.130(c)(2) and (7), which includes, but is not limited to January of each calendar year.
For each of these points in time CMS reviews the applicable data to determine whether a generic drug is approved or a biosimilar is licensed and such generic drug or biosimilar is subject to Bona Fide Marketing (which includes prior to selection for negotiation or renegotiation of selected drugs, after selection of drugs for negotiation or renegotiation, or after a selected drug ceases to be a selected drug), beginning with drug selection for negotiation and renegotiation for initial price applicability year 2029 and initial price applicability years thereafter, we are proposing a schedule for reviewing the data at proposed § 429.130(c) that varies from the policies in the applicable guidance for drugs that were potentially eligible for selection or have been selected for initial price applicability years 2026, 2027, and 2028. We have observed across the administration of the Negotiation Program to date that there are certain points in time across a calendar year that are key to the drug selection process of the Negotiation Program and/or have an impact on CMS and manufacturer operations related to plan year coverage contracts and related determinations. For example, it is necessary for CMS to review whether any applicable generic drugs and biosimilars are subject to Bona Fide Marketing as part of the review steps CMS undertakes to identify qualifying single source drugs for purposes of drug selection for negotiation (proposed at § 429.130(c)(1)) and to identify selected drugs ineligible to be selected for renegotiation (proposed at § 429.130(c)(3)) prior to the selected drug publication date. Once a drug is selected, but prior to the agreement of an MFP or the end of the negotiation period during the negotiation process, CMS must regularly review if any applicable generic drugs and biosimilars are subject to Bona Fide Marketing as an essential part of determining if a selected drug remains subject to the negotiation process. Therefore, consistent with the policies for implementation as described in Negotiation Program Guidance, CMS would maintain monthly reviews of data during this period as proposed at § 429.130(c)(4) for drugs selected for
( printed page 36266)
negotiation and as proposed at § 429.130(c)(5) for drugs selected for renegotiation (if any) for initial price applicability years 2029 and thereafter. However, after the negotiation period ends, we believe that less frequent reviews are sufficient and would provide manufacturers, Part D plan sponsors, and other interested parties with notice regarding the specific points in time across a calendar year at which CMS would review the specified data and provide public notice of any selected drugs that are deselected (consistent with proposed § 429.135(a) and (d)) because CMS determined that a generic drug is approved or a biosimilar is licensed for the selected drug and such generic drug or biosimilar is subject to Bona Fide Marketing. For example, we understand that manufacturers, plan sponsors, and other relevant parties may begin negotiations for Part D plan bids more than a year prior to the plan year and CMS' deadline for Part D sponsor bids (see § 423.265(b)). Therefore, we believe that one key date includes October to ensure manufacturers and Part D plan sponsors are aware prior to the subsequent June due date for Part D plan sponsors to provide CMS with any required plan bids for the next Medicare plan contract year (as indicated in § 423.265(b)(1)) of any selected drugs that may be deselected. For example, if we determine in October 2028 that a generic drug or biosimilar is approved or licensed, as applicable, for a selected drug for initial price applicability year 2029 and that generic drug or biosimilar is subject to Bona Fide Marketing, that selected drug ceases to be a selected drug on January 1, 2030 and the MFP would not be applicable for that year. Manufacturers and plan sponsors would then be aware in November 2028 that the selected drug would cease to be a selected drug in contract year 2030, bids for which are due in June 2029. Finally, as described in proposed § 429.130(c)(2) and (7), we would periodically monitor, including at least annually in January prior to each selected drug publication date, the marketing of any applicable generics and biosimilars in the following circumstances: (1) if a potential qualifying single source drug was determined by CMS not to qualify as a qualifying single source drug for any prior initial price applicability year because CMS determined that at least one generic drug was approved using the potential qualifying single source drug as the listed drug or at least one biosimilar was licensed using the potential qualifying source drug as the reference product, and such generic drug or biosimilar was subject to Bona Fide Marketing, and (2) if a selected drug ceases to be a selected drug in accordance with proposed § 429.135(a) (in other words, because CMS determined that a generic drug is approved or a biosimilar is licensed for the selected drug and such generic drug or biosimilar is subject to Bona Fide Marketing).
For awareness for manufacturers and other interested parties, we propose in § 429.130(a)(1)(i) through (iv) that CMS would review the last 12 months or the four quarters of data, as applicable, ending with the last full month or quarter of data available to CMS at the time of its review. Specifically, for PDE data, consistent with proposed § 429.130(a)(1)(i) and using the example of CMS' review in October, this means that CMS would review PDE data reported from October of the prior calendar year through September of the current calendar year. Submission deadlines for the data source would determine the data available to CMS at the time of review. For example, AMP and ASP data are submitted to CMS by manufacturers on a monthly or quarterly basis in accordance with regulations (42 CFR 447.510 and 42 CFR 414.805(a)(5)), whereas PDE data for selected drugs are submitted to CMS by Part D plan sponsors within 7 days from the date the Part D plan sponsor receives the claim for selected drugs or within 30 days for all other drugs (per 42 CFR 423.325). CMS would pull such data in the month CMS reviews such data, and the specific date on which CMS may pull the data would vary year to year and may be impacted, for example, by the date of Federal holidays.
Consistent with the policies for implementation as described in Negotiation Program Guidance, we provide the following examples of potential factors considered in CMS' review of whether an approved generic drug or licensed biosimilar is subject to Bona Fide Marketing. While the circumstances illustrated in these examples weigh in favor of, or against, considering a generic drug or biosimilar to be subject to Bona Fide Marketing, CMS' inquiry for any particular drug would be based on the totality of the circumstances and not on the presence, or absence, of any single factor. First, as an example, if a potential qualifying single source drug has at least one approved generic drug or licensed biosimilar that has high and consistent PDE utilization, AMP sales, and/or ASP sales, we would consider the generic(s) or biosimilar(s) of the potential qualifying single source drug to be subject to Bona Fide Marketing. As a second example, a potential qualifying single source drug might have a newly or recently approved generic drug or licensed biosimilar and the product has relatively low PDE utilization, AMP sales, and/or ASP sales. In this example, if CMS finds in additional review of public information that the generic drug or biosimilar manufacturer has successfully launched their product, and there is no evidence of agreements limiting distribution of the generic drug or biosimilar, then we may consider the generic drug or biosimilar of the potential qualifying single source drug to be subject to Bona Fide Marketing. As a third example, a potential qualifying single source drug might have an approved generic drug or licensed biosimilar product with no PDE utilization, AMP sales, and/or ASP sales. In this example, if CMS finds in additional review of public information that there are ongoing patent disputes and no generic drug or biosimilar manufacturer has successfully launched their product, then we may consider the generic drug or biosimilar of the potential qualifying single source drug as not subject to Bona Fide Marketing.
As discussed in section I.A.2. of this proposed rule, because sections 11001(c) and 11002(c) of the IRA require CMS to implement the Negotiation Program provisions in sections 11001 and 11002 of the IRA for 2026, 2027, and 2028 through program instruction or other forms of program guidance, prior to January 1, 2029, the requirements and processes, including with respect to Bona Fide Marketing and deselection of a selected drug, for a selected drug that was included on the list of selected drugs with respect to initial price applicability year 2026, 2027, or 2028 are set forth with respect to such years in the applicable program guidance. Revisions to the implementation of such policies for 2026, 2027, and 2028 with respect to drugs selected for initial price applicability years 2026, 2027, and 2028 would be addressed by CMS through publication of revised guidance. In accordance with the expiration of the statutory program instruction requirement at the end of 2028, we propose in section I.A.2. of this proposed rule that the provisions of this proposed rule, including the policies set forth in §§ 429.130 and 429.135 with respect to Bona Fide Marketing and deselection of a selected drug, as applicable, would apply starting in 2029 with respect to the drugs selected for initial price applicability years of 2026, 2027, or 2028.
Section 1192(c)(1) of the Act specifies that each negotiation-eligible drug
( printed page 36267)
included on the list of drugs published for an initial price applicability year consistent with section 1192(a) of the Act will be a “selected drug” with respect to that year and each subsequent year beginning before the first year that begins at least 9 months after the date on which the Secretary determines at least one drug is approved under section 505(j) of the FD&C Act using the selected drug as the listed drug or at least one biological product is licensed under section 351(k) of the PHS Act using the selected drug as the reference product, and that such drug or biological product is marketed pursuant to such approval or licensure. Additionally, section 1192(c)(2) of the Act states that a negotiation-eligible drug shall not be subject to the negotiation process under section 1194 of the Act for the applicable negotiation period but shall continue to be considered a selected drug for purposes of the number of negotiation-eligible drugs published on the list with respect to such initial price applicability year if the Secretary determines that the criteria in section 1192(c)(1) of the Act are met before or during the negotiation period for that initial price applicability year. Therefore, in accordance with sections 1192(c)(1) and (2) of the Act, we propose in § 429.135 to codify provisions related to deselection of a selected drug subject to the timeline and situations proposed in § 429.135(b).
Specifically in proposed § 429.135(a), each drug selected for negotiation for an initial price applicability year would remain a selected drug, with respect to such initial price applicability year and each subsequent year beginning before the first year that begins at least 9 months after the date on which CMS determines that at least one generic drug is approved under section 505(j) of the FD&C Act using any dosage form or strength of the selected drug as the listed drug or at least one biosimilar is licensed under section 351(k) of the PHS Act using any dosage form or strength of the selected drug as the reference product, and that such generic drug or biosimilar is subject to Bona Fide Marketing.[44]
First, we would use FDA reference sources, including the Orange Book and Purple Book, to determine whether a generic drug or biosimilar is approved or licensed for any strength(s) or dosage form(s) of a selected drug as proposed in § 429.135(a)(1). Second, if we determine that a generic drug or biosimilar has been approved or licensed, we would consider whether such generic drug or biosimilar is subject to Bona Fide Marketing as proposed in 429.130(a) and consistent with proposed § 429.135(a)(2). For clarity, we note that a selected drug remains a selected drug unless and until it is deselected as proposed in § 429.135(a) without regard to whether the Primary Manufacturer decides to execute a Negotiation Program Agreement as proposed in § 429.200, to terminate a Negotiation Program Agreement as proposed in § 429.205, or to transfer the Negotiation Program Agreement to a new Primary Manufacturer as proposed in § 429.210.
Proposed § 429.135(b) clarifies that the circumstances described in § 429.135(b) would apply to such selected drug based on the date as of which we determine the conditions described in § 429.135(a) are met. As detailed in proposed § 429.135(b)(1), if we determine on a date during the period beginning on the selected drug publication date (as defined in § 429.20) and ending on November 1 of the year that begins 2 years prior to the initial price applicability year for which the drug was selected for negotiation, that a selected drug has a generic drug that is approved or biosimilar that is licensed and such generic drug or biosimilar is subject to Bona Fide Marketing, pursuant to section 1192(c)(2) of the Act, the selected drug ceases to be subject to the negotiation process under section 1194 of the Act; an MFP would not be published for, or apply to, such drug; and the selected drug would remain a selected drug only for that initial price applicability year but would not be replaced by another selected drug. As discussed in proposed § 429.130(c)(7), after CMS determines a selected drug would cease to be a selected drug consistent with § 429.135(a), CMS would monitor whether the generic drug or biosimilar for the selected drug continues to be subject to Bona Fide Marketing.
If CMS makes such determination that a selected drug ceases to be a selected drug (consistent with the criteria in proposed § 429.135(a)) on a date during the period of time beginning on November 2 of the year that begins 2 years prior to the initial price applicability year for which the drug was selected for negotiation and ending on March 31 of that initial price applicability year, then the selected drug would cease to be a selected drug on January 1 of the year following the initial price applicability year for which such drug was selected for negotiation and the MFP would apply only for the initial price applicability year for which such drug was selected for negotiation (consistent with proposed § 429.135(b)(3)).
If the date CMS makes such a determination is during the selected drug's price applicability period after March 31 of the initial price applicability year for which the selected drug is selected for negotiation, then the selected drug would cease to be a selected drug on January 1 of the year that begins at least 9 months after the date that CMS determines the conditions are met (consistent with proposed § 429.135(a)) and the MFP would apply until such date that the selected drug ceases to be a selected drug (consistent with proposed § 429.135(b)(4)).
These different scenarios for the timing of deselection in accordance with proposed § 429.135(b) are summarized in Table 1 using a drug selected for negotiation for initial price applicability year 2029 as an illustrative example.
Table 1—Deselection of a Selected Drug Following Generic Drug or Biosimilar Approval/Licensure and Marketing
Date on which CMS determines that a generic drug or biosimilar is approved/licensed and marketed
Result with respect to selected drug for the negotiation program
The selected drug publication date for initial price applicability year 2029 through November 1, 2027 (the end of the Negotiation Period for initial price applicability year 2029)
Selected drug remains a selected drug for initial price applicability year 2029, though MFP does not apply; selected drug ceases to be a selected drug on January 1, 2030.
( printed page 36268)
November 2, 2027 through March 31, 2029
Selected drug remains a selected drug and MFP applies for initial price applicability year 2029; selected drug ceases to be a selected drug on January 1, 2030.
April 1, 2029 through March 31, 2030
Selected drug remains a selected drug and MFP applies for initial price applicability year 2029 and calendar year 2030; selected drug ceases to be a selected drug on January 1, 2031.
April 1, 2030 through March 31, 2031
Selected drug remains a selected drug and MFP applies for initial price applicability year 2029 and calendar years 2030 and 2031; selected drug ceases to be a selected drug on January 1, 2032.
April 1, 2031 through March 31, 2032
Selected drug remains a selected drug and MFP applies for initial price applicability year 2029 and calendar years 2030, 2031 and 2032; selected drug ceases to be a selected drug on January 1, 2033.
As we propose at § 429.135(c), if CMS determines a generic drug is approved or biosimilar is licensed for a selected drug, and such generic drug or biosimilar is subject to Bona Fide Marketing prior to the selected drug publication date for the next initial price applicability year (including drugs previously selected for renegotiation), a drug selected for a prior initial price applicability year is not eligible for renegotiation for such next initial price applicability year consistent with section 1194(f)(5) of the Act.
C. Negotiation Program Agreement (§§ 429.200 Through 429.210)
1. Entrance Into an Agreement With CMS (§ 429.200)
Section 1193(a) of the Act directs CMS to enter into agreements with manufacturers of selected drugs with respect to a price applicability period, by not later than February 28 following the selected drug publication date and provides for certain requirements of such agreements. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 40.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
In section 1191(c)(1) of the Act, the Negotiation Program statute adopts the definition of “manufacturer” established in section 1847A(c)(6)(A) of the Act. Section 1193(a)(1) of the Act establishes that CMS will negotiate, or renegotiate, as applicable, an MFP with “the manufacturer” of the selected drug. To the extent that more than one entity meets the definition of manufacturer for a selected drug for purposes of initial price applicability year 2029 and subsequent years, we would designate the entity that holds the NDA(s)/BLA(s) for the selected drug to be “the manufacturer” of the selected drug, hereinafter referred to as the “Primary Manufacturer” (as defined in proposed § 429.20). We would refer to any other entity that meets the statutory definition of manufacturer for a drug product included in the selected drug and that either: (1) is listed as a manufacturer in an NDA or BLA for the selected drug; or (2) markets the selected drug pursuant to an agreement with the Primary Manufacturer but is not listed on the NDA or BLA, as a Secondary Manufacturer (as defined in proposed § 429.20). Consistent with sections 1193(a)(4) and 1193(a)(5) of the Act, and as described in this proposed part 429, the Primary Manufacturer of a selected drug that enters into a Negotiation Program Agreement must collect and report all necessary information applicable to the selected drug, inclusive of NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer. Likewise, as the entity that is party to the Negotiation Program Agreement, the Primary Manufacturer will be solely responsible for compliance with all provisions of the Negotiation Program Agreement and will be accountable for ensuring compliance with respect to units of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer.
With respect to initial price applicability year 2029 and subsequent years, consistent with the policies for implementation as described in section 40.1 of the Negotiation Program Guidance, proposed § 429.200 establishes the Negotiation Program Agreement and the requirements of such which a willing Primary Manufacturer is subject to upon executing a Negotiation Program Agreement with CMS. We would not enter into a Negotiation Program Agreement with any Secondary Manufacturer of a selected drug with respect to that selected drug. In accordance with section 1193(a) of the Act, as proposed in § 429.200(a)(1), the deadline for a Primary Manufacturer to enter into a Negotiation Program Agreement is 11:59 p.m. Pacific Standard Time (PST) on February 28 following the selected drug publication date with respect to the initial price applicability year for which the selected drug was selected for negotiation. We propose at § 429.200(a)(2) that the negotiation period would begin on the earlier of two dates: (1) February 28 of the year of the selected drug publication date with respect to the initial price applicability year for which the selected drug was selected for negotiation; or, (2) the date that the Negotiation Program Agreement is fully executed by 11:59 p.m. PST. The Negotiation Program Agreement would be executed on the day that the last party to sign the Negotiation Program Agreement signs the Negotiation Program Agreement and would be in effect until terminated in accordance with proposed § 429.205(a).
We have established a model Negotiation Program Agreement in accordance with section 1193(a) of the Act. A Primary Manufacturer's obligation to make the MFP available applies during the entirety of the selected drug's price applicability period for which the manufacturer has a Negotiation Program Agreement in place. At proposed § 429.200(b), consistent with the provisions of such model Negotiation Program Agreement, we propose to codify requirements that a willing Primary Manufacturer will agree to comply with upon execution of the Negotiation Program Agreement, namely: (1) to comply with all applicable requirements and conditions set forth in sections 1191 through 1198 of the Act and all applicable guidance
( printed page 36269)
and regulations, including in part 429, implementing those provisions and any changes to the Act that affect the Negotiation Program; (2) to negotiate to determine an MFP for the selected drug with CMS, during the negotiation period for the initial price applicability year for the selected drug, in accordance with section 1194 of the Act, including as described in subpart F of this part (as described in II.F. of this proposed rule); (3) as applicable, to renegotiate to determine an MFP for the drug selected for renegotiation with CMS, during the renegotiation period for the initial price applicability year for the drug selected for renegotiation, in accordance with section 1194 of the Act, including as described in subpart G of this part (as described in II.G. of this proposed rule); (4) to provide access to the MFP, including as renegotiated, with respect to the selected drug, during the selected drug's price applicability period, in accordance with section 1193(a)(3), including as described in subpart B and subpart I of this part (as described in II.B. and II.I. of this proposed rule); (5) to submit to CMS, in a form and manner specified by CMS, the information specified in sections 1191 to 1198 of the Act, the Negotiation Program Agreement, or this part, including but not limited to, the information as specified at §§ 429.100(d), 429.405, 429.505(b), and, if applicable, § 429.615(b)(1) (as described in sections II.B.1, II.E.2, II.F.2, and if applicable, section II.G.4. of this proposed rule), in accordance with sections 1193(a)(4) and 1193(a)(5) of the Act; and (6) to comply with requirements determined by CMS to be necessary for the purposes of administering the Negotiation Program and monitoring compliance with the Negotiation Program, in accordance with section 1193(a)(5) of the Act, including as described in subpart J of this part (as described in section II.J. of this proposed rule).
Consistent with policies for implementation as described in section 40.1 of the Negotiation Program Guidance, we propose in § 429.200(c) that the Negotiation Program Agreement must be signed by an authorized representative of the Primary Manufacturer as defined in proposed § 429.20 and by CMS. In § 429.20, we propose that an authorized representative of the Primary Manufacturer must be the Primary Manufacturer's Chief Executive Officer (CEO), Chief Financial Officer (CFO), an individual with equivalent authority to a CEO or CFO, or an individual that has been granted delegation of signature authority on behalf of one of these categories of individuals. The authorized representative(s) must be legally authorized to bind the Primary Manufacturer to the terms and conditions contained in the Negotiation Program Agreement, including any Addenda. CMS would specify a form and manner to access and sign the Negotiation Program Agreement as indicated in proposed § 429.200(c). Consistent with prior initial price applicability years, we intend to continue to use the CMS Health Plan Management System (“the CMS HPMS”) for electronic access and signing of the Negotiation Program Agreement. Specifically, to make a request to obtain electronic signature access via the CMS HPMS, an authorized representative must prepare an official letter that states the user's name(s), role(s) (for example, Chief Executive Officer), CMS user ID, the P number that would be used for CMS and the Primary Manufacturer to interact for the purposes of the Negotiation Program related to the relevant selected drug, and that electronic signature access is required. We note that it is a requirement of the CMS HPMS that the person accessing the CMS HPMS have a Social Security Number (SSN), among other requirements, to meet the identity proofing requirements for system access.
Consistent with policies for implementation as described in section 40.1 of the Negotiation Program Guidance, we propose in § 429.200(d) that the Negotiation Program Agreement will take effect on the date that the Negotiation Program Agreement is signed both by an authorized representative of the Primary Manufacturer and by CMS. The term of the Negotiation Program Agreement shall be from the effective date until the Negotiation Program Agreement is terminated in accordance with § 429.205(a). As proposed in § 429.200(e), the Primary Manufacturer and CMS will formalize the agreement to an MFP through the execution of an Addendum to the Negotiation Program Agreement. The Addendum to the Negotiation Program Agreement will be made available to the Primary Manufacturer in a form and manner specified by CMS and must be signed by an authorized representative of the Primary Manufacturer and CMS. Instructions for completing the Negotiation Program Agreement and a template of the Negotiation Program Agreement are available on the CMS website.[45]
2. Termination (§ 429.205)
Section 1193(b) of the Act requires that, when the Primary Manufacturer enters into the Negotiation Program Agreement described in proposed § 429.200, the Negotiation Program Agreement will remain in effect, including through renegotiation, as applicable, until the selected drug is no longer a selected drug consistent with CMS' determination in accordance with section 1192(c) of the Act (as described in proposed § 429.135 and section II.B.7.of this proposed rule). With respect to 2026 through 2028, CMS implemented these requirements in section 40.6 of the Negotiation Program Guidance. With respect to initial price applicability year 2029 and subsequent years, consistent with the policies for implementation as described in section 40.6 of the Negotiation Program Guidance, we are proposing at § 429.205 that when the Primary Manufacturer enters into the Negotiation Program Agreement described in proposed § 429.200, the Negotiation Program Agreement will remain in effect, including through renegotiation, as applicable, until the selected drug is no longer a selected drug consistent with CMS' determination in accordance with section 1192(c) of the Act (as described in proposed § 429.135 and section II.B.7. of this proposed rule) unless the Negotiation Program Agreement is terminated sooner by the Primary Manufacturer under the process established in proposed § 429.205(b).
Participation in the Negotiation Program is voluntary. And so, in accordance with section 1193(a)(5) of the Act, a Primary Manufacturer may terminate its Negotiation Program Agreement with respect to a selected drug with respect to a price applicability period, before reaching an agreement with CMS as to the MFP for the selected drug or after such an MFP is agreed to, if the Primary Manufacturer meets certain conditions for termination consistent with the provisions in 26 U.S.C. 5000D(c). Specifically, a Primary Manufacturer seeking to terminate its Negotiation Program Agreement with respect to a selected drug must submit to CMS a Request to Terminate, as defined in proposed § 429.20, that meets all requirements established in proposed § 429.205(b)(1). Section 11003 of the IRA expressly connects a Primary Manufacturer's financial responsibilities under the voluntary Negotiation Program to that manufacturer's voluntary participation in the Medicaid
( printed page 36270)
Drug Rebate Program, the Coverage Gap Discount Program,[46]
and the Manufacturer Discount Program. The provisions enacted in 26 U.S.C. 5000D give the Primary Manufacturer choices with regard to the Negotiation Program. One option is that the Primary Manufacturer may participate in the Negotiation Program. Another option is that the Primary Manufacturer may opt out of the Negotiation Program, and the excise tax may be imposed under 26 U.S.C. 5000D. Alternatively, the Primary Manufacturer may opt out of the Negotiation Program but avoid the excise tax on sales of the selected drug during periods for which the Primary Manufacturer does not have applicable program agreements, as defined in proposed § 429.20, with the aforementioned Medicare and Medicaid programs and none of its drugs are covered by an agreement under the Manufacturer Discount Program under section 1860D-14C of the Act. Promoting continuity in the administration of the Negotiation Program warrants extending parallel options to a Primary Manufacturer with respect to potential civil monetary penalty liability, as described in proposed subpart K and section II.C.10. of this proposed rule. A Primary Manufacturer with a Negotiation Program Agreement with respect to the price applicability period with respect to a selected drug may opt out of the Negotiation Program and pay civil money penalties associated with violations of program requirements. Alternatively, a Primary Manufacturer seeking to cease participation in the Negotiation Program through the end of the price applicability period for a selected drug may avoid civil monetary penalty liability by terminating its Negotiation Program Agreement if it also ceases participation in the Medicaid Drug Rebate Program and the Manufacturer Discount Program through the end of the price applicability period for the selected drug.
In proposed § 429.205(a)(1) and (a)(2), we propose the processes by which termination from the Negotiation Program may occur. Specifically, we propose that a Negotiation Program Agreement will terminate effective as of the date of the earlier of: (1) The first date that the selected drug covered by the Negotiation Program Agreement is no longer a selected drug consistent with CMS's determination in accordance with section 1192(c) of the Act as described at § 429.135; or (2) The date of termination established in proposed § 429.205(b)(5) in connection with a Request to Terminate by the Primary Manufacturer submitted under proposed § 429.205(b), that is, absent a Primary Manufacturer request to rescind its Request to Terminate in accordance with proposed § 429.205(d), the first date following the receipt of a Request to Terminate that CMS determines none of the drugs of the Primary Manufacturer are covered by an agreement under the Manufacturer Discount Program in accordance with 42 CFR 423.2752(c)(1).
In proposed § 429.205(b)(1), in accordance with section 1193(a)(5) of the Act, CMS proposes that a Primary Manufacturer that wishes to terminate a Negotiation Program Agreement may submit in writing a Request to Terminate, in a form and manner specified by CMS, which must include both: (1) a request for termination of the Primary Manufacturer's applicable program agreements under the Medicaid Drug Rebate Program and the Manufacturer Discount Program, consistent with the requirements as set forth in 26 U.S.C. 5000D(c)(1)(A)(i); and, (2) an attestation that through the end of the price applicability period for the selected drug, the Primary Manufacturer: (i) shall not seek to enter into any subsequent applicable program agreement; and (ii) shall not seek coverage for any of its drugs under the Manufacturer Discount Program under section 1860D-14C of the Act, consistent with the requirements as set forth in 26 U.S.C. 5000D(c)(1)(B). In proposed § 429.205(f), CMS proposes that a Primary Manufacturer that terminates in accordance with proposed § 429.205(b) and later seeks to re-enter any applicable program agreement or obtain coverage for any of its drugs under the Manufacturer Discount Program during the selected drug's price applicability period would be deemed to have provided an invalid attestation that was a condition of termination, and the Negotiation Program Agreement would once again become operative as of the date of re-entry into the applicable program agreement(s) or coverage for any of the Primary Manufacturer's drugs under the Manufacturer Discount Program. If a Primary Manufacturer terminated its Negotiation Program Agreement pursuant to the process described at § 429.205(b)(1) prior to completing the negotiation process and agreeing to an MFP, and such Primary Manufacturer later sought to re-enter any applicable program agreement or obtain coverage for any of its drugs under the Manufacturer Discount Program, the negotiation process would be initiated or resumed in accordance with the negotiation process proposed in subpart F and section II.C.6. of this proposed rule. In addition, the timing of the Primary Manufacturer's decision to resume participation in the Negotiation Program may implicate the renegotiation process described in proposed subpart G and section II.C.7. of this proposed rule.
We propose in § 429.205(b)(2) that if the Primary Manufacturer's notice of termination contains all required elements under proposed § 429.205(b)(1), CMS will confirm receipt of the Primary Manufacturer's notice and execute the actions with respect to termination of the Primary Manufacturer's applicable program agreements as described in the proposed § 429.205(b)(3), (b)(4), and (b)(5). As noted previously, section 11003 of the IRA expressly connects a Primary Manufacturer's financial responsibilities under the voluntary Negotiation Program to that manufacturer's voluntary participation in the Medicaid Drug Rebate Program and the Manufacturer Discount Program. As described in proposed § 429.205(b)(3), if a Primary Manufacturer determines after executing its Negotiation Program Agreement that it is unwilling to continue its participation in the Negotiation Program and provides a Request to Terminate that CMS determines complies with the requirements in proposed § 429.205(b)(1), the Primary Manufacturer's Request to Terminate will constitute good cause for CMS to terminate the Primary Manufacturer's applicable program agreement(s) under the Manufacturer Discount Program, as applicable, in accordance with section 1860D-14C(b)(4)(B)(i) of the Act and 42 CFR 423.2752(c)(1) of this chapter, and to expedite the date on which none of the drugs of the Primary Manufacturer are covered by an agreement under the Manufacturer Discount Program in accordance with 42 CFR 423.2752(c)(1). CMS will automatically grant a Request to Terminate that complies with the requirements in proposed § 429.205(b)(1). Consistent with section 1860D-14C(b)(4)(B)(i) of the Act and 42 CFR 423.2752(c)(1) of this chapter, a termination of a Manufacturer Discount Program agreement by CMS will not be effective earlier than 30 calendar days
( printed page 36271)
after the date of notice from CMS to the manufacturer of such termination after CMS determines that the Primary Manufacturer's Request to Terminate complies with proposed § 429.205(b)(1). CMS has proposed codifying the established deadlines for the Negotiation Program in proposed subpart F, including the date by which CMS will provide the final offer to Primary Manufacturers, to provide a Primary Manufacturer with sufficient time to ensure all conditions for termination are met by the end of the negotiation period set forth in section 1191(b)(4)(B) of the Act.
As described in proposed § 429.205(b)(4), if CMS determines the Primary Manufacturer's Request to Terminate complies with the requirements in proposed § 429.205(b)(1), the Request to Terminate also will constitute good cause for CMS to terminate the Primary Manufacturer's applicable program agreement(s) under the Medicaid Drug Rebate Program in accordance with section 1927(b)(4)(B)(i) of the Act and the Medicaid National Drug Rebate Agreement (NDRA). CMS will automatically grant a Request to Terminate that complies with the requirements in proposed § 429.205(b)(1). The Primary Manufacturer's applicable program agreements include all the NDRAs of the Primary Manufacturer. Consistent with section 1927(b)(4)(B)(i) of the Act and the NDRA, such termination by CMS will not be effective earlier than 60 days after the date of CMS' notice to the Primary Manufacturer of such termination.
As discussed previously, in the proposed § 429.205(b)(5), unless a Primary Manufacturer rescinds its Request to Terminate in accordance with the proposed § 429.205(d), CMS will terminate the Negotiation Program Agreement effective on the first date following the receipt of a Request to Terminate that CMS determines complies with the requirements in proposed § 429.205(b)(1) on which none of the drugs of the Primary Manufacturer are covered by an agreement under the Manufacturer Discount Program in accordance with 42 CFR 423.2752(c)(1).
Consistent with policies as described in section 40.1 of the Negotiation Program Guidance, § 429.205(c) proposes the process by which a Primary Manufacturer may decide not to execute a Negotiation Program Agreement with CMS and expedite its exit from the Manufacturer Discount Program to meet the conditions of 26 U.S.C. 5000D(c). As described in proposed § 429.200(a)(2), the negotiation period will begin on the earlier of two dates: (1) February 28 of the year of the selected drug publication date; or, (2) the date that the Negotiation Program Agreement is fully executed. If a Negotiation Program Agreement is not fully executed by February 28 following the publication of the selected drug list, a period will begin on March 1 of the year in which the selected drug list is published, during which the Primary Manufacturer could be exposed to potential excise tax liability, in accordance with 26 U.S.C. 5000D(b)(1). Section 429.205(c) proposes that a Primary Manufacturer that does not wish to participate in the Negotiation Program and that seeks to avoid potential excise tax liability may submit to CMS in writing a Request to Terminate, in a form and manner specified by CMS, that meets the requirements described in proposed § 429.205(b)(1), consistent with the requirements set forth in 26 U.S.C. 5000D(c). In response to such a Request to Terminate, we propose to take the steps described in § 429.205(b)(2) through (5), as applicable.
If a Primary Manufacturer decides not to execute a Negotiation Program Agreement with CMS, and follows the process proposed at § 429.205(c), and later enters into applicable program agreements or obtains coverage under the Manufacturer Discount Program for any of its drugs, but never executes a Negotiation Program Agreement, the tax suspension period described in 26 U.S.C. 5000D(c) will end. See 26 U.S.C. 5000D(c)(1)(B).
Section 429.205(d) proposes the process by which a Primary Manufacturer may rescind the Primary Manufacturer's Request to Terminate submitted pursuant to proposed § 429.205(b) or (c). A Primary Manufacturer may request to rescind the Request to Terminate by submitting to CMS a written request for a hearing, in a form and manner specified by CMS. If a Primary Manufacturer provides a Request to Terminate that complies with the requirements in proposed § 429.205(b)(1), in accordance with section 1860D-14C(b)(4)(B)(i) of the Act, upon written request to rescind from such Primary Manufacturer, we propose to provide a hearing concerning termination of the Primary Manufacturer's applicable program agreements under the Manufacturer Discount Program. Such a hearing would be held prior to the effective date of termination of the applicable program agreements with sufficient time for such effective date to be repealed. Such a hearing would be held solely on the papers; because CMS' determination that there is good cause for termination depends solely on the Primary Manufacturer's request for termination to effectuate its decision not to participate in the Negotiation Program, the only question to be decided in the hearing is whether the Primary Manufacturer has asked to rescind its termination request prior to the effective date of the termination. CMS intends to automatically grant a request from the Primary Manufacturer to rescind its Request to Terminate.
Likewise, as proposed at § 429.205(d), if a Primary Manufacturer submits a Request to Terminate that CMS determines complies with the requirements in proposed § 429.205(b)(1), in accordance with section 1927(b)(4)(B)(i) of the Act, upon written request to rescind from such Primary Manufacturer, we propose to provide a hearing concerning termination of the Primary Manufacturer's applicable NDRA(s) under the Medicaid Drug Rebate Program. Such a hearing would be held prior to the effective date of termination of the applicable program agreements with sufficient time for such effective date to be repealed. Such a hearing would be held solely on the papers; because CMS' determination that there is good cause for termination depends solely on the Primary Manufacturer's request for termination to effectuate its decision not to participate in the Negotiation Program, the only question to be decided in the hearing is whether the Primary Manufacturer has asked to rescind its termination request prior to the effective date of the termination. We intend to automatically grant a request from the Primary Manufacturer to rescind its Request to Terminate. If a Primary Manufacturer's request to rescind termination of the applicable program agreements under the Manufacturer Discount Program and the Medicaid Drug Rebate Program is granted, the Primary Manufacturer's Request to Terminate will also be rescinded.
In proposed § 429.205(e), CMS proposes requirements about the effect of termination and the Primary Manufacturer's continuing responsibilities under the Negotiation Program Agreement. That is, we propose that notwithstanding any termination of the Negotiation Program Agreement for a selected drug in accordance with either § 429.205(a)(1) or (a)(2), the Primary Manufacturer would continue to be responsible for making the MFP for the selected drug available, in accordance with subpart I, with respect to any unit of the selected drug
( printed page 36272)
dispensed, administered, or furnished prior to the effective date of termination under § 429.205(a). Also, we propose that notwithstanding the termination of the Negotiation Program Agreement, any confidentiality, record retention, or data requirements and any requirements for Primary Manufacturer participation in audit and other Negotiation Program oversight activities shall continue to apply.
3. Other Provisions of the Negotiation Program Agreement (§ 429.210)
With respect to initial price applicability year 2029 and subsequent years, consistent with policies for implementation as described in section 40.7 of the Negotiation Program Guidance, we propose at § 429.210(a) to codify that if any provision of the Negotiation Program Agreement is found to be invalid by a court of law with competent jurisdiction, the Negotiation Program Agreement would be construed in all respects as if any invalid or unenforceable provision(s) were eliminated, and without any effect on any other provision. Further, as proposed at § 429.210(b), CMS retains the authority to amend the Negotiation Program Agreement to reflect changes in law, regulations, or guidance as applicable. When possible, CMS would give the Primary Manufacturer at least 60 days of notice of any change to the Negotiation Program Agreement.
In accordance with section 1193(a)(5) of the Act, consistent with policies for implementation as described in section 40.7 of the Negotiation Program Guidance, we propose at § 429.210(c) to codify that if, after entering in a Negotiation Program Agreement with CMS, the Primary Manufacturer of a selected drug transfers ownership of one or more NDAs or BLAs, as applicable, of a selected drug to another entity, the Primary Manufacturer remains responsible for all requirements of the Negotiation Program Agreement associated with the transferred NDA(s) or BLA(s), including the requirement to provide access to the MFP as described in proposed subpart I, unless and until the Primary Manufacturer transfers all the NDAs or BLAs of the selected drug that it holds to an entity and such acquiring entity assumes responsibility as the new Primary Manufacturer. CMS proposes in § 429.210(c)(1)(ii) that the acquiring entity's assumption of responsibility as the new Primary Manufacturer must be evidenced by a novation to the transferring Primary Manufacturer's original Negotiation Program Agreement, which, as discussed below, must be provided to CMS for review and approval at least 30 calendar days before the intended effective date of the proposed transfer. In proposed § 429.210(c)(1)(ii), CMS also requires that the novation be signed by the transferring Primary Manufacturer and the acquiring entity and must include, at minimum, the legal name of the acquiring entity (which would be the new Primary Manufacturer), the effective date of the transfer of ownership of all transferred NDAs or BLAs of the selected drug that the transferring Primary Manufacturer holds and of the transfer of responsibility for all requirements of the Negotiation Program Agreement to the acquiring entity, a list of all transferring NDAs or BLAs of the selected drug, and agreement that the acquiring entity assumes all obligations and liabilities under the transferring Primary Manufacturer's Negotiation Program Agreement as the successor in interest.
We propose at § 429.210(c)(2), that if the transferring Primary Manufacturer submits a novation agreement that meets the requirements proposed at § 429.210(c)(1)(ii), and is approved by CMS, the acquiring entity becomes the successor in interest to the transferring Primary Manufacturer's Negotiation Program Agreement and the Primary Manufacturer of the applicable selected drug as of the novation's effective date of the transfer of responsibility for all requirements of the Negotiation Program Agreement.
We propose at § 429.210(c)(1)(ii), that the transferring Primary Manufacturer must provide CMS with documentation of the intended transfer of responsibility for all requirements of the Negotiation Program Agreement to the acquiring entity, in the form of a novation, at least 30 calendar days before the intended effective date of any such transfer, for CMS review and approval. We encourage Primary Manufacturers to consult with CMS regarding their potential novation before submission of the novation to CMS at least 30 calendar days before the intended effective date of any such transfer.
We propose at § 429.210(c)(3) that the transferring Primary Manufacturer remains responsible for any outstanding Negotiation Program rebate liabilities related to the Biosimilar Delay under section 1192(f) of the Act unless, and until, such liabilities are transferred to the acquiring entity as the new Primary Manufacturer.
If the Primary Manufacturer of a selected drug transfers all NDAs or BLAs of the selected drug, and the acquiring entity assumes responsibility as the new Primary Manufacturer of the selected drug for purposes of the Negotiation Program, we recognize that this transfer of ownership could enable the original Primary Manufacturer to avoid potential excise tax liability for future sales, as well as render unnecessary the efforts by the original Primary Manufacturer to comply with the statutory suspension of the excise tax and the termination process for a Primary Manufacturer seeking to invoke the statutory suspension of the excise tax. We recognize that whether this transfer of ownership would have these impacts may depend on whether the transfer of the NDA(s) or BLA(s) was made to an entity that is not a related party and complied with relevant principles of tax law.
D. Program Administration (§ 429.300)
1. Confidentiality Policy and Data Use (§ 429.300)
Section 1193(c) of the Act directs CMS to consider manufacturer-submitted information that CMS deems proprietary to only be used by CMS or disclosed to and used by the Comptroller General of the United States for purposes of carrying out the Negotiation Program. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 40.2.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
We are proposing at § 429.300(a) to codify the requirement that Primary Manufacturer submitted information that CMS deems proprietary would only be used by CMS or disclosed to and used by the Comptroller General of the United States for purposes of carrying out the Negotiation Program and we are proposing at § 429.300(b) to establish a confidentiality policy that would deem proprietary information, as described in proposed § 429.300(a), including trade secrets and confidential commercial or financial information, as confidential information exempt from disclosure if the information meets the requirements set forth under Exemption 3 or Exemption 4 of the Freedom of Information Act (5 U.S.C. 552(b)(3), (4)). We would not disclose PHI or PII, except in accordance with applicable laws, where received by CMS as proposed in §§ 429.505 and 429.615 or information received by CMS in engagement with interested parties specified in §§ 429.515 and 429.620(e), as proposed in § 429.300(b)(1).
We are proposing at § 429.300(c) to codify certain data elements submitted by a Primary Manufacturer of a selected drug proposed in §§ 429.405 and 429.505(b), or of a drug selected for
( printed page 36273)
renegotiation proposed in § 429.615(b)(1), as applicable, that shall be considered confidential information of the Primary or Secondary Manufacturer and would be deemed proprietary information by CMS, unless the information is publicly available. We propose at § 429.300(c)(1) through 429.300(c)(5) that CMS would consider non-FAMP and associated non-FAMP data collection, research and development costs of the Primary Manufacturer for the selected drug, current unit costs of production and distribution of the selected drug, data on pending patent applications for the selected drug, and market data and revenue and sales volume for the selected drug in the United States as proprietary, unless the information that is provided to CMS is already publicly available, in which case it would be considered non-proprietary. We would consider prior Federal financial support, approved patent information, exclusivities, and approved applications under section 505(c) of the FD&C Act or section 351(a) of the PHS Act that are publicly available to be non-proprietary. Information submitted by a Primary Manufacturer to CMS on a particular data element as a part of the data submission in proposed §§ 429.405, 429.505, and 429.615, as applicable, and described in sections II.E.2., II.F.2., and II.G.4.b. of this proposed rule (which CMS anticipates collecting via the Drug Price Negotiation ICR) may include information that is non-proprietary because it is publicly available or does not represent trade secrets and confidential commercial or financial information, such as the introductory language within an explanation field of a data element. Additionally, we propose in § 429.300(c)(6) that if a Primary Manufacturer submits a Common Technical File/Drug Master File/“drug dossier” as a part of their submission, in accordance with proposed § 429.505(d)(3) or § 429.615(b)(1)(i) (if applicable), we would consider this to be proprietary information.
Finally, as a part of the Drug Price Negotiation ICR for data submissions in proposed §§ 429.505(b)(2) and (d)(3), and 429.615(b), as applicable, a Primary Manufacturer may indicate for CMS which information the Primary Manufacturer believes should be withheld from disclosure by CMS consistent with existing Federal requirements for protecting proprietary information, including Exemptions 3 and/or 4 of the FOIA, and that are not included under proposed § 429.300(c) as data that must be deemed proprietary information by CMS.
A Primary Manufacturer may choose to publicly disclose information regarding its ongoing negotiations with CMS at its discretion. If a Primary Manufacturer discloses information that is made public regarding any aspect of the negotiation process prior to the explanation of the MFP being released by CMS, we reserve the right to publicly discuss the specifics of the negotiation process regarding that Primary Manufacturer. CMS would not publicly discuss ongoing negotiations with a Primary Manufacturer, otherwise. If a Primary Manufacturer chooses to disclose any material that is made public that CMS has previously deemed to be proprietary information of that Primary Manufacturer, CMS would no longer consider that specific material proprietary consistent with proposed § 429.300.
As proposed in § 429.705(b) (as described in section II.H.2. of this proposed rule), we propose to make public the explanation for the MFP, which includes: the narrative explanation of the MFP; redacted information regarding the negotiation meetings, as applicable, including exchanges of offers and counteroffers, as applicable; and the redacted information submitted by a Primary Manufacturer in proposed in § 429.505(b)(2) or § 429.615(b)(1) (as described in section II.F.2. or II.G.4. of this proposed rule), as applicable, and the redacted information submitted by interested parties in proposed § 429.505(d)(3) or § 429.615(b)(2), as applicable. We propose at § 429.300(d) that CMS must redact any information deemed proprietary or covered by the confidentiality policy described in § 429.300(a) and 429.300(b), including the publication of the MFP. The narrative explanation of the negotiation process would not include any information that CMS deemed to be proprietary information of the Primary Manufacturer, PHI or PII (defined in proposed § 429.20), or information that is protected from disclosure under other applicable law that may have been submitted through a Primary Manufacturer's data submission or an individual's data submission. In advance of this public narrative, CMS may share certain aggregate or non-selected drug specific information, for example regarding status of the negotiation process or conclusion of negotiations without sharing any information that CMS deemed to be proprietary information of the Primary Manufacturer, PHI, PII, or information that is protected from disclosure under other applicable law. If the Primary Manufacturer chooses to disclose proprietary information prior to the explanation of the MFP, then it would not be redacted in the explanation of the MFP.
Within the explanation of the MFP, CMS may also make public high-level comments about the data submitted to CMS, as proposed in § 429.405 and proposed in §§ 429.505 or 429.615 (as applicable) that are determined to be proprietary, without sharing any PHI/PII or any proprietary information reported to CMS under section 1193(a)(4) of the Act for purposes of the negotiation. For example, CMS will not make public the research and development costs reported by a Primary Manufacturer, as CMS would treat that data as proprietary, but CMS may say “the manufacturer has recouped its research and development costs.” Any proprietary information obtained during an audit will also remain confidential, except as necessary to use that information in the course of a judicial enforcement proceeding.
As proposed in § 429.515(a)(5) and described in section II.F.4.a. of this proposed rule, CMS would prohibit audio or video recording of any negotiation meetings between CMS and a Primary Manufacturer. We would maintain written records of the negotiation process, including negotiation meetings, in compliance with applicable Federal law, including the Federal Managers Financial Integrity Act and the Federal Records Act. A Primary Manufacturer may maintain its own written record of the negotiation process.
We would retain all records pertaining to the Negotiation Program, including but not limited to records submitted by a Primary Manufacturer, in compliance with the Federal Records Act, as implemented by the CMS Policy for Records and Information Management.[47]
Data submitted by Primary Manufacturers, as described in proposed in §§ 429.405, 429.505, and 429.615 (as applicable) would be classified as Bucket 3: financial records in the CMS Records Schedule and would be managed in accordance with the CMS Records schedule.[48]
( printed page 36274)
E. Establishment of a Single MFP and Determination of the Ceiling (§§ 429.400 Through 429.445)
1. Establishment of a Single MFP for Negotiation and Renegotiation Purposes (§ 429.400)
Section 1191(c)(3) of the Act states that an MFP means, with respect to a year during a price applicability period and with respect to a selected drug with respect to such period, the price negotiated under section 1194 of the Act, and updated under section 1195(b) of the Act, as applicable, for such drug and year. We interpret this language to refer to negotiation of a selected drug with respect to the price applicability period for that selected drug. Consistent with respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including for example, with respect to initial price applicability year 2028, section 60.1 of the Negotiation Program Guidance. Consistent with the policies for implementation as described in section 60.1 of the Negotiation Program Guidance, we propose in § 429.400 to identify a single price for each selected drug, including for selected drugs with multiple dosage forms and strengths. This single price would be used at each step in the negotiation process, as described in proposed §§ 429.510 through 429.535 and sections II.F.3. through II.F.8. of this proposed rule, and for each step in the renegotiation process, as described in proposed § 429.620 and section II.G.5. of this proposed rule. Accordingly, each offer and counteroffer, as described in proposed §§ 429.520 through 429.535 for negotiation and § 429.620(f) through (i) for renegotiation, would include a single price, including for a selected drug with multiple dosage forms, strengths, and formulations. We also propose in § 429.400(a)(1) to base the single price on the cost of the selected drug per 30-day equivalent supply (rather than per unit—such as tablet, capsule, injection—or per volume or weight-based metric) for all formulations (including drugs payable under Part B, covered under Part D, or both, as applicable), weighted across dosage forms and strengths for the purposes of determining a single price included in an initial offer proposed in §§ 429.510(d) and § 429.520 and as described in sections II.F.3.c. through II.F.3.d. and section II.F.5. of this proposed rule and for the purpose of additional offers during the negotiation as proposed in §§ 429.525 through 429.535 and as described in sections II.F.4. and II.F.6. through II.F.8. of this proposed rule. This approach of negotiating a single price across all dosage forms and strengths based on a 30-day equivalent supply of the selected drug both aligns with the statutory requirement at section 1194(a)(1) of the Act to negotiate a single MFP for each selected drug and would allow for a more direct comparison between the selected drug and its therapeutic alternative(s) (described in proposed § 429.510(b)), if any, which might also have different dosage forms, strengths, and treatment regimens (for example, daily consumption of tablets versus monthly injections of solutions) when developing the initial offer. The 30-day equivalent supply methodology is proposed in § 429.415 and described in section II.E.4. of this proposed rule.
2. Collection of Non-FAMP (§ 429.405)
Section 1193(a)(4)(A) of the Act establishes that a Primary Manufacturer that has entered into a Negotiation Program Agreement with CMS (proposed in § 429.200 and described in section II.C.1. of this proposed rule), in accordance with the requirements of its Negotiation Program Agreement, is required to submit, for the negotiation period for the price applicability period, and, if applicable, before any period of renegotiation, information on the non-FAMP (as defined in 38 U.S.C. 8126(h)(5)) for the selected drug for the applicable year or period. Section 1194(c)(1)(C)(ii) of the Act, with respect to a selected drug selected for initial price applicability year of 2027 or later, provides that the MFP negotiated for the selected drug shall not exceed the lower of the amount calculated pursuant to section 1194(c)(1)(B) of the Act or section 1194(c)(1)(C) of the Act, where the calculation pursuant to section 1194(c)(1)(C) of the Act requires identifying the lower of: (1) the average non-FAMP price for the selected drug for 2021 (or, in the case that there is not an average non-FAMP available for such selected drug for calendar year 2021, for the first full calendar year following the market entry for such drug), as adjusted for inflation; or (2) the average non-FAMP for the selected drug for the calendar year prior to the selected drug publication date for the selected drug. Section 1194(c)(6) of the Act further provides that the term “average non-Federal average manufacturer price” means the average of the non-Federal average manufacturer price (as defined in 38 U.S.C. 8126(h)(5)) for the four calendar quarters of the year involved.
With respect to initial price applicability years 2027 and 2028, CMS implemented these requirements through guidance, including, for example, with respect to initial price applicability year 2028, section 50.1.1 of the Negotiation Program Guidance. Consistent with the policies implemented through section 50.1.1 of the Negotiation Program Guidance, we are proposing at § 429.405(a) to codify the requirement that, in accordance with the requirements of its Negotiation Program Agreement, a Primary Manufacturer of a selected drug must submit to CMS, in a form and manner specified by the agency, by 11:59 p.m. PST on March 1 of the calendar year of the selected drug publication date with respect to the initial price applicability year for which the selected drug was selected for negotiation, inclusive of NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer, the non-FAMP, unit type, and total unit volume for each NDC-11 of the selected drug for all quarters of the four quarters of calendar year 2021 in which the selected drug was sold and non-FAMP data was reported (or, in the case that there is not an average non-FAMP available for such selected drug for calendar year 2021, the non-FAMP, unit type, and total unit volume for each NDC-11 of the selected drug for the four quarters of the first full calendar year following the market entry for such drug), and for the calendar year prior to the selected drug publication date with respect to the initial price applicability year for which the selected drug, was selected for negotiation. This requirement to submit information is not inclusive of NDC-11s of the selected drug that are not manufactured, marketed, controlled, or sold by the Primary Manufacturer or a Secondary Manufacturer, or NDC-11s to which the MFP would not apply, such as sample packages, inner packages, and discontinued products. We intend to consider the average non-FAMP to be available for a selected drug for calendar year 2021 if non-FAMP data has been reported for at least one NDC-11 of the selected drug for at least one quarter in calendar year 2021. For a given NDC-11 of a selected drug, when there are at least 30 days of commercial sales data but less than a calendar quarter of data to calculate the non-FAMP in calendar year 2021 (or the first full year following market entry of such drug, when applicable) or the calendar year prior to the selected drug publication date for the selected drug, the non-FAMP reported by the Primary Manufacturer to CMS for that calendar quarter should reflect the temporary non-FAMP predicated upon the first 30 days of commercial sales data. The temporary non-FAMP should be calculated
( printed page 36275)
following the same methodology used to calculate the temporary non-FAMP amount used to determine the Temporary Federal Ceiling Price, as described in the Department of Veterans Affairs (VA) 2025 Updated Guidance for Calculation of Federal Ceiling Prices (FCPs) for New Drugs subject to Public Law 102-585 (including any restatements of the non-FAMP made in any manufacturer non-FAMP submissions to the VA).
In accordance with sections 1193(a)(4)(A), 1194(b)(2)(A), 1194(c)(1)(C)(ii), 1194(c)(6), and 1193(a)(5) of the Act, as described in proposed § 429.405(a)(1), we propose that this data collection would be mandatory for Primary Manufacturers and would be due by March 1 of the year of the selected drug publication date with respect to the initial price applicability year for which the selected drug was selected for negotiation. This due date also aligns with the due date for submission of information related to the factors listed at section 1194(e) of the Act (further described in proposed § 429.505 and II.F.2. of this proposed rule). We intend to collect the non-FAMP through an Information Collection Request (ICR), which will include additional parameters for this data collection.
Additionally, in accordance with section 1193(a)(5) of the Act, we propose at § 429.405(b) that if the non-FAMP is restated due to requirements of 38 U.S.C. 8126 and implementing regulations and guidance issued by the VA, then in accordance with the requirements of its Negotiation Program Agreement as set forth in § 429.200, the Primary Manufacturer is required to update the submission of non-FAMP to CMS for the selected drug.
3. Determination of the Ceiling (§ 429.410)
a. Limitations on Offer Amount (§ 429.410(a))
Section 1194(b)(2)(F) of the Act states the limitations on an offer amount and requires that in negotiating the MFP of a selected drug, including during renegotiation, we will not make an offer or agree to a counteroffer for an MFP that exceeds the ceiling as determined in section 1194(c) of the Act or, if applicable, is less than the Temporary Floor for Small Biotech Drugs as determined in section 1194(d) of the Act. Consistent with section 60.2.1 of Negotiation Program Guidance, we propose at § 429.410(a) to codify the requirement that CMS would not make an offer or agree to a counteroffer for an MFP that exceeds the ceiling amount under section 1194(c) of the Act. We also propose at § 429.440(a) that CMS would not make an offer or agree to a counteroffer for an MFP that is less than the Temporary Floor for Small Biotech Drugs, if applicable.
b. Determination of the Ceiling (§ 429.410(b))
Section 1194(c)(1)(A) of the Act states that the MFP negotiated for a selected drug, with respect to the first initial price applicability year of the price applicability period with respect to such drug, shall not exceed the lower of the amount under section 1194(c)(1)(B) of the Act or the amount under section 1194(c)(1)(C) of the Act. In section 60.2.1 of the Negotiation Program Guidance, CMS developed a process to calculate the amounts described in section 1194(c)(1)(B) and (c)(1)(C) of the Act. We propose to codify that process at § 429.410(b). Provisions regarding the ceiling calculation for drugs selected for renegotiation are proposed at § 429.620(b) and discussed at section II.G.5.a. of this proposed rule. The process we propose to codify at § 429.410(b) provides for a single ceiling amount to apply for each selected drug, in accordance with the references in sections 1194(b)(2)(F) and 1194(c)(1)(A) of the Act to “the ceiling” or “the lower of the amount under subparagraph (B) or the amount under subparagraph (c),” respectively, both in the singular.
In proposed § 429.410(b)(1), we propose to identify the amount under section 1194(c)(1)(B) of the Act, consistent with section 60.2.1 of the Negotiation Program Guidance, as one of the following amounts, as applicable to the selected drug:
As described in proposed § 429.410(b)(1)(i), for a selected drug that is covered under Part D but is not payable under Part B, the sum of the plan-specific enrollment weighted amounts, described in section 1194(c)(1)(B)(i) of the Act and as determined under proposed § 429.420 and discussed in section II.E.5. of this proposed rule.
As described in proposed § 429.410(b)(1)(ii), for a selected drug that is payable under Part B and paid according to section 1847A(b)(4) of the Act, but is not covered under Part D, the payment amount under section 1847A(b)(4) of the Act for the year prior to the year of that selected drug's publication date with respect to the initial price applicability year for that selected drug, described in section 1194(c)(1)(B)(ii) of the Act and as determined under proposed § 429.425 and discussed in section II.E.6. of this proposed rule.
As described in proposed § 429.412(b)(1)(iii), for a selected drug that is payable under Part B and paid according to section 1847A(b)(4) of the Act, and covered under Part D, an amount equal to the combined Part B and Part D amount, as determined under proposed § 429.430 and discussed in section II.E.7. of this proposed rule.
As described in proposed § 429.410(b)(1)(iv), for a selected drug that is payable under Part B but is not paid according to section 1847A(b)(4) of the Act,[49]
and is covered under Part D, an amount based on the sum of the plan-specific enrollment weighted amounts, described in section 1194(c)(1)(B)(i) of the Act and as determined under proposed § 429.420 and discussed in section II.E.5. of this proposed rule.
As described in proposed § 429.410(b)(1)(v), for a selected drug that is payable under Part B but is not paid according to section 1847A(b)(4) of the Act, and is not covered under Part D, there is no amount as determined under proposed paragraph § 429.410(b)(1).
The previous two bullet points describe scenarios where a selected drug is payable under Part B but is not paid according to the methodology provided in section 1847A(b)(4) of the Act. Since such selected drugs would not have a “payment amount” determined under such section, we propose that the ceiling amount described under section 1194(c)(1)(B)(ii) of the Act would not be applicable to the determination because no such amount exists. In such instances, we propose to calculate and consider the other ceiling amounts specified under section 1194(c)(1) of the Act for such drugs, as applicable. When such a selected drug is payable under Part B but not paid according to section 1847A(b)(4) of the Act and is covered under Part D, we propose to use the sum of the plan-specific enrollment weighted amount. However, for a selected drug that is payable under Part B but not paid according to section 1847A(b)(4) of the Act and is not covered under Part D, there would be no applicable ceiling amount described in section 1194(c)(1)(B) of the Act, and so we propose to use only the applicable percent of average non-FAMP described in section 1194(c)(1)(C) of the Act and determined as proposed in § 429.410(b)(2) to calculate the ceiling.
( printed page 36276)
Section 1194(c)(1)(C)(ii) of the Act requires that for initial price applicability year 2027 and subsequent years, the lower of two different average non-FAMP amounts described in sections 1194(c)(1)(C)(ii)(I) and 1194(c)(1)(C)(ii)(II) of the Act be used as the amount described under section 1194(c)(1)(C) of the Act. CMS implemented these requirements through guidance, including, for example, section 60.2.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028. We propose in § 429.410(b)(2), that the applicable percent, as applicable to the selected drug, will be the lower of the average non-FAMP amounts described in section 1194(c)(6) of the Act. We will compare the amounts described in § 429.410(b)(2) to determine which is lower and serve as the payment amount to compare the amount described in § 429.410(b)(1). In accordance with 1194(c)(1)(A) of the Act, the selected drug's ceiling is the lower of the amount determined under § 429.410(b)(1) or (b)(2).
The amounts determined under proposed § 429.410(b)(2) would be the lower of the average non-FAMP for each selected drug for calendar year 2021 (or in the case that there is not an average non-FAMP for such drug for calendar year 2021, for the first full year following the market entry for such drug), increased by the percentage increase in the CPI-U from September 2021 (or December of such first full year following the market entry), as applicable, to September of the year that is 3 years prior to the initial price applicability year of the selected drug proposed in § 429.410(b)(2)(i) and the average non-FAMP for such selected drug for the calendar year prior to the selected drug publication date proposed in § 429.410(b)(2)(ii). Regarding the first referenced amount, we interpret section 1194(c)(1)(C)(i) of the Act to indicate that if there is no non-FAMP for any of the NDCs for the selected drug for calendar year 2021, then we would use the first full year following market entry of the selected drug. Similarly, regarding the second referenced amount, in a situation where none of the NDCs for the selected drug are reported in the calendar year prior to the selected drug publication date, we would then apply the average non-FAMP reported for such NDCs from the first full year prior to the year of the selected drug publication date.
We propose at § 429.410(b)(3), to calculate a single amount, a 30-day equivalent supply as described in § 429.415, across all dosage forms and strengths of the selected drug for the amounts calculated in §§ 429.420 through 429.435 to determine which of these amounts is the lowest and would serve as the ceiling for the MFP for the selected drug. We solicit comments on recommended approaches for CMS to our proposals throughout this section, including calculation of the 30-day equivalent supply, the determination of the different ceiling amounts, and the temporary floor for small biotech drugs.
4. Calculation of the 30-Day Equivalent Supply (§ 429.415)
Consistent with the policies for implementation as described in section 60.2.1.1 of the Negotiation Program Guidance, we propose at § 429.415 a methodology for determining the 30-day equivalent supply for a selected drug and therapeutic alternative(s), as applicable. The price per 30-day equivalent supply methodology facilitates the determination of a single ceiling price, and the negotiation of a single MFP, across dosage forms and strengths of a selected drug where units (for example, milligrams versus milliliters) and treatment regimens (for example, daily consumption of tablets versus monthly injections of solutions) differ. The proposed 30-day equivalent supply methodology also provides a standardized methodology to calculate a price for a standardized treatment duration for all dosage forms and strengths of selected drugs and its therapeutic alternative(s), if any; gives a more complete picture of beneficiary utilization when compared to a unit-based approach; and allows for comparison across the selected drug and the respective therapeutic alternative(s), if any.
We considered several alternatives, including those submitted through public comment for the Negotiation Program Guidance for initial price applicability year 2028. The alternatives that we considered included a per-unit approach; price per course of treatment; consulting with the Primary Manufacturer of each selected drug to determine a methodology; and a flexible methodology to accommodate the clinical disease state and treatment landscape. We do not believe these alternatives provide for the standardization needed across different formulations, dosage forms, and strengths of a selected drug and its therapeutic alternative(s), if any, necessary to consider the selected drug as described in section 1194(e) of the Act and apply a single ceiling and a single MFP across dosage forms and strengths of the selected drug as required in section 1191(c)(3) of the Act. In addition, using a different methodology for selected drugs that are covered under Part D from selected drugs that are payable under Part B or for selected drugs that are both covered under Part D and payable under Part B could lead to inconsistencies and issues with comparing the selected drug to therapeutic alternative(s), if any.
Using a per-unit price approach, particularly for drugs payable under Part B, would not provide the standardization necessary to compare a selected drug with a therapeutic alternative nor to apply a single ceiling and a single MFP for a selected drug. This is because for drugs payable under Part B, unit type may differ both across HCPCS codes within a selected drug and across therapeutic alternatives payable under Part B, especially if a drug is both payable under Part B and covered under Part D. Using a price-per-course of treatment approach to creating a 30-day equivalent supply is time and resource intensive because treatment duration and dosing may be specific to individual patients and can also differ due to factors such as individual adherence, delays due to adverse events, or expected gaps due to adjuvant therapy or procedures. The option wherein we would work with the Primary Manufacturer on an individual methodology for each selected drug would be time and resource intensive and potentially not lead to a standardized approach and methodology that could be used for selected drugs and its therapeutic alternative(s), if any. This approach would also create inconsistency across selected drugs. We also believe that a flexible 30-day equivalent supply methodology to accommodate the clinical disease state and treatment landscape, wherein we would use FDA labeling and treatment guidelines to tailor the methodology, would not capture patient-specific dosing regimens whereas the proposed approach using days between services approach would capture such patient-specific variation. variation.
After considering these alternatives, we propose at § 429.415(a)(1), that for selected drugs that are covered under Part D, we would use the methodology as described at 42 CFR 423.104(d)(2)(iv)(A)(2) which relies on the “days' supply” field in PDE records to calculate the 30-day equivalent supply for each PDE record associated with the selected drug.
Consistent with the policies for implementation as described in section 60.2.1.1 of the Negotiation Program Guidance, we propose at § 429.415(a)(2) that CMS will use a different methodology to calculate the 30-day
( printed page 36277)
equivalent supply for selected drugs payable under Part B because Part B data does not contain a “days' supply” field like the PDE records for selected drugs that are covered under Part D. The methodology we propose to use for calculating the 30-day equivalent supply for drugs payable under Part B involves calculating a “days between services” amount for each instance of Part B data associated with the selected drug. As an example of the approach proposed at § 429.415(a)(2)(ii), consider if the date of service for the first instance of Part B data is January 12, 2025, and the immediately subsequent instance of Part B data with the same active moiety/active ingredient has a date of service of March 12, 2025, then the first claim or record's “days between services” amount would be calculated as 59 days. If the beneficiary's third instance of Part B data was August 1, 2025, the number of “days between services” amount, for days between March 12, 2025, and August 1, 2025, would be calculated as 142 days for the second claim or record. The resulting “days between services” amounts in this example would be the same amount if the subsequent claim was a PDE record rather than an instance of Part B data. We propose at § 429.415(a)(2)(i)(B) that the subsequent instance of Part B data or any PDE record be for a drug or biological product with the same active moiety/active ingredient identified as set forth at § 429.125(b), as the selected drug.[50]
We propose at § 429.415(a)(2)(ii)(A) if the beneficiary has an instance Part B data that does not have a subsequent instance of Part B data or PDE record with the same active moiety/active ingredient, identified as set forth at § 429.125(b), CMS would assign a “days between services” amount equal to the median “days between services” amount for all prior instances of Part B claims or records or PDE records for that selected drug associated with that beneficiary during the applicable claims period. Thus, if the beneficiary's last instance of Part B data was August 1, 2025, and there is no subsequent instance of Part B data or PDE record of the same active moiety/active ingredient, it would be assigned a median of 59 days and 142 days, which is 100.5 days. We propose at § 429.415(a)(2)(ii)(B) that if there is only one instance of Part B data for the selected drug and there are no other instances of Part B data or PDE records with the same active moiety/active ingredient associated with the beneficiary during the applicable claims period then we would not assign a “days between services” amount and furthermore not include the single instance of Part B data in the calculation of 30-day equivalent supply. The proposal at § 429.415(a)(2)(ii)(B) would not apply for drugs typically administered one time because there would most likely never be subsequent instances of Part B data. We discuss our proposed approach for drugs typically administered one time later in this section.
We understand that drugs payable under Part B often have recurrent administrations and variable dosing depending on body weight, indication, treatment phase, or response to treatment. The proposed “days between services” methodology addresses these concerns by using observed data from individual patients, which can potentially capture if providers are using patient-specific dosing regimens which can cause the variations. The “days between services” methodology would reduce the possibility of undervaluing the costs of drugs payable under Part B when attempting to determine the cost per 30-day equivalent supply.
Part B data is billed at the HCPCS code level, and some calculations using the 30-day equivalent supply of the selected drug require NDC-level 30-day equivalent supply amounts, so we propose at § 429.415(a)(2)(iv) to allocate a portion of the 30-day equivalent supply calculated at the level of each HCPCS code to each NDC within the HCPCS code that includes a selected drug to calculate a 30-day equivalent supply at the NDC level. We propose at § 429.415(a)(2)(v) steps to determine the total Part B 30-day equivalent supply at the NDC level.
We propose at § 429.415(a)(2)(v)(E)
(2)
to assign a value of “12” for the 30-day equivalent supply for drugs that are typically administered one time (for example, some vaccines and cancer therapies), by identifying HCPCS codes that generally have a median of one claim per calendar year. This value would apply to any selected drug and therapeutic alternative(s) of the selected drug, if any, for purposes of determining the initial offer as described in § 429.510, that are typically administered one time and payable under Part B. However, unlike for claims described in § 429.415(a)(2)(ii)(B), the reason for a lack of a second claim for these drugs is because these drugs are typically administered once for the lifetime of a patient. A “days between services” amount would therefore generally not be calculated for any claim associated with such drugs under the proposed methodology at § 429.415(a)(2)(ii) without this second claim. To calculate a single amount for the ceiling and for offers and counteroffers for the MFP under the methodologies proposed under this subpart E, a value for the 30-day equivalent supply needs to be assigned for these drugs in order to be included in the calculation. We propose that assigning a value of “12” as a 30-day equivalent supply facilitates the necessary calculations in a manner that reasonably and accurately reflects such drugs usage. This proposal would also be consistent with our policy of developing a standardized methodology to calculate a price for all dosage forms and strengths of selected drugs and its therapeutic alternative(s).
We considered an alternative approach to the proposed 30-day equivalent supply methodology for drugs typically administered one time described previously, wherein we would use a price-per-administration or price per HCPCS billing unit for such drugs. Under this alternative approach, instead of calculating a 30-day equivalent supply and price per 30-day equivalent supply, we would instead calculate a price-per-administration or price-per-HCPCS billing unit for a selected drug. However, if such selected drug includes multiple HCPCS codes, calculating a single price per administration across HCPCS codes may not be appropriate (for example, if the existing price-per-administration differs significantly across HCPCS codes due to different treatment patterns). Calculating a price-per-administration would also not account for different volumes of the drug used in different instances of administration, which would conflict with our typical use of a payment limit per HCPCS billing unit for payment under Part B.
5. Determination of the Sum of the Plan-Specific Enrollment Weighted Amounts (§ 429.420)
Section 1194(c)(2) of the Act describes the calculation for the sum of the plan-specific enrollment weighted amount for prescription drug plan or an MA-PD plan with respect to a covered Part D drug for purposes of section 1194(c)(1)(B)(i) of the Act. Consistent with the policies for implementation as described in section 60.2.2.1 of the Negotiation Program Guidance, we propose at § 429.420(b) to use a 30-day equivalent supply methodology to calculate the sum of the plan-specific enrollment weighted amounts.
( printed page 36278)
Plan sponsors report Part D PDE data to CMS at the NDC-11 level and report direct and indirect remuneration data to CMS at the NDC-11 level in the annual Detailed Direct and Indirect Remuneration (DIR) Report. As directed by statute, and proposed at § 429.420(a) and (b)(1), we propose to use plan sponsors' reported Part D PDE data and DIR data for the year that is 4 years prior to the initial price applicability year of the selected drug, which would be the most recent year for which data would be available, for purposes of determining the sum of the plan-specific enrollment weighted amounts for selected drugs that are covered under Part D. As proposed at § 429.420(a), we would include all Part D plans found in the PDE data that meet the inclusion criteria described in the definition of “total expenditures under Part D” at proposed § 429.20. We intend to identify Part D plans based on the combination of the Part D contract identifier and the plan benefit package identifier.
We propose at § 429.420(a)(1) to use the list of NDC-11s of the selected drug (as determined in proposed § 429.100(c) and discussed in section II.B.1. of this proposed rule), to determine which NDC-11s of the selected drug are included in this ceiling calculation.
We note that because we would not have PDE data for Part D plans in the following circumstances, such Part D plans would, by definition, be excluded from the calculation proposed at § 429.420(b):
Plans that have no utilization of the selected drug; and
Plans that have no enrollment for the year that is 4 years prior to the initial price applicability year of the selected drug.[51]
We propose at § 429.420(b)(1) to use the most recent year for which all data is available, which is generally the year that is 4 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation. We propose at § 429.420(b)(2) for each Part D plan and each NDC-11 we would sum the total direct and indirect remuneration amounts found in the DIR Report for the year that is 4 years prior to the initial price applicability year of the selected drug and subtract the total ERPOSA calculated in proposed § 429.420(b)(3) to avoid double counting price concessions applied at the point of sale. The amount calculated under proposed § 429.420(b)(4) is the NDC-11 price per unit, net of all price concessions received by such Part D plan or pharmacy benefit manager on behalf of such Part D plan. To determine which month(s) of enrollment to include in our analysis, we conducted an analysis of monthly Part D plan enrollment changes during 2022 and determined that monthly enrollment changes were the lowest from November to December, so we propose at § 429.420(b)(5) that December would be the most stable month to identify Part D plan enrollment. The choice of one month to identify enrollment, rather than an average annual enrollment, also allows the weights calculated at proposed § 429.420(b)(6) to sum to one.
6. Determination of the Payment Amount Under Section 1847A(b)(4) of the Act (§ 429.425)
Section 1194(c)(1)(B)(ii) of the Act provides for the use of an amount equal to the payment amount under section 1847A(b)(4) for a selected drug that is payable under Part B for the year prior to the year of the selected drug publication date with respect to the initial price applicability year for the selected drug. Consistent with the policies for implementation as described in section 60.2.2.2 of the Negotiation Program Guidance and in accordance with section 1194(c)(1)(B)(ii) of the Act, CMS proposes at § 429.425 to calculate for a selected drug payable under Part B and paid under section 1847A(b)(4) of the Act, an amount equal to the payment amount under section 1847A(b)(4) of the Act for the year prior to the year of the selected drug publication date with respect to the initial price applicability year for that drug or biological product (that is, 3 years prior to the initial price applicability year of the selected drug). We interpret “payment amount under section 1847A(b)(4)” under section 1194(c)(1)(B)(ii) of the Act to refer to an amount calculated for such year calculated using the methodology specified in section 1847A(b)(4) of the Act which is, for single source drugs and biological products, separately calculating the annual ASP and WAC described at proposed § 429.425(a)(2)(i) and taking the lesser of, as proposed at § 429.425(a)(2)(ii). When calculating this amount, we propose at § 429.425(a)(1) to use the list of NDC-11s of the selected drug (as determined in proposed § 429.100(c) and discussed in section II.B.1. of this proposed rule).
Additionally, as directed by statute, we propose to calculate the payment amount under section 1847A(b)(4) of the Act by using the quarterly reported ASP and WAC data for the calendar year 3 years prior to the initial price applicability year of the selected drug, for the purpose of determining the payment amount under section 1847A(b)(4) of the Act. This calculated amount proposed in § 429.425(a)(2) would apply for only selected drugs that are payable under Part B and paid according to section 1847A(b)(4) of the Act. When calculating the payment amount under 1847A of the Act for each HCPCS code we would use NDC-level data from the ASP Data Collection System (that is, the ASP portal). ASP data are required to be submitted to the ASP portal within 30 days after the close of each calendar quarter, and CMS uses that data to calculate payment amounts under section 1847A of the Act for the subsequent quarter, which results in a two-quarter lag between the quarter of ASP data used and when the Part B payment limits apply. We would apply this two-quarter lag when conducting all relevant calculations of the ceiling and application of MFP that rely on data from the ASP portal. For example, when calculating the annual ASP to determine the “payment amount under section 1847A(b)(4)” of the Act for calendar year 2028, CMS would use ASP portal data reported for the third quarter of 2027 through the second quarter of 2028.
To calculate a payment amount under section 1847A(b)(4) of the Act as described in section 1194(c)(1)(B)(ii) of the Act for a 30-day equivalent supply across all dosage forms and strengths of a selected drug, we propose at § 429.425(a)(2) to calculate such amount for each HCPCS code to which NDC-11s of the selected drug are assigned using data from manufacturers of all NDC-11s that are assigned to the HCPCS code and are part of the selected drug, and then assign such payment amount to each NDC-11 of the selected drug within such HCPCS code. We propose at § 429.425(a)(3) to allocate HCPCS code-level utilization from Part B data across each NDC-11 assigned to such HCPCS code, so that we may then use the NDC-level utilization as weights when calculating a single payment amount under section 1847A(b)(4) of the Act, across all dosage forms and strengths of the selected drug. We would achieve this allocation by using the proportion of ASP units reported by manufacturers to CMS for each NDC-11 that is assigned to the HCPCS code.
We propose at § 429.425(a)(2)(i) to convert the quarterly ASP and WAC reported by the manufacturer for the year as set forth in § 429.425(a) for each NDC-11 for a selected drug that is associated with a HCPCS code to an
( printed page 36279)
annual calendar year ASP or WAC amount for each HCPCS code. We propose to do so by taking an average of the reported ASP or WAC amounts for the HCPCS code across all four quarters of such calendar year, weighted by the total number of billing units in the Part B data within that HCPCS code each quarter.
We propose at § 429.425(a)(2)(i)(A), that if the total number of billing units in the Part B data within the HCPCS code are zero for a given quarter, we would assign that quarter the lowest positive total units from among the other quarters in the same calendar year for that HCPCS code.
We propose at § 429.425(a)(2)(i)(B), that for each of the separate ASP and WAC calculations, if the reported price is negative, zero, or missing for all applicable NDC-11s assigned to the HCPCS code in a given quarter for the calendar year as set forth in § 429.405(a), we would exclude that quarter from the applicable calculation.
We propose at § 429.425(a)(2)(i)(C), that if the WAC reported to the ASP portal is negative, zero, or missing for all applicable NDC-11s assigned to the HCPCS code for all four quarters for the calendar year as set forth in § 429.405(a), then we would use the WAC reported by the Primary Manufacturer of the selected drug to CMS under section 1194(e)(1) of the Act as described in § 429.505(b)(2)(v).
We propose at § 429.425(a)(2)(iii), if ASP, WAC reported to the ASP payer portal, and WAC reported by the Primary Manufacturer pursuant to section 1194(e)(1) of the Act as described in § 429.505(b)(2) are all negative, zero, or missing for all applicable NDC-11s assigned to the HCPCS code for all four quarters of the calendar year as set forth in § 429.405(a), we would calculate the payment amount under section 1847A(b)(4) of the Act by taking the average of the published payment limits in the ASP pricing file (or in the OPPS Addendum B file, if it is not available in the ASP pricing file) for the HCPCS code across all four quarters, weighted by the total number of billing units in the Part B data for that HCPCS code for the four quarters of the calendar year as set forth in § 429.415(a)(2)(i) (including the adjustments made when billing units are zero). We believe this approach is appropriate because the prices in these files are typically calculated following the methodology described in section 1847A(b)(4) of the Act but may be adjusted as necessary to accommodate the underlying data (for example, negative, zero, or missing ASP and/or WAC). This amount would apply to all applicable NDC-11s assigned to the associated HCPCS code.
We propose at § 429.425(a)(2)(ii) that we would use the lesser of the annual ASP and the annual WAC as determined under proposed § 429.425(a)(2)(i) to yield the payment amount under section 1847A(b)(4) of the Act for the associated HCPCS code. As proposed at § 429.425(a)(1)(iv), we propose to exclude from the determination of the payment amount under section 1847A(b)(4) NDC-11s of the selected drug that are self-administered drugs. Self-administered drugs that are adjudicated through pharmacy claims are not used in the CMS calculation of the payment amounts under section 1847A(b)(4) of the Act for drugs that are not selected for negotiation. Therefore, we believe the exclusion of self-administered drugs from the determination of the payment amount for selected drugs under section 1847A(b)(4) of the Act is appropriate since self-administered drugs are not factored into payment amounts under section 1847A(b)(4) for drugs that are not selected for negotiation.
We propose at § 429.425(a)(3) the steps for allocating HCPCS code-level utilization from Part B data for Part B services. For the steps proposed at § 429.425(a)(3)(ii), we have streamlined the steps given our experiences from previous initial price applicability years, specifically initial price applicability year 2028. The units, measured as the Part B data converted to the NCPDP level units that are used in PDE records, are used in cases where we need to sum together the Part D quantity dispensed and the Part B quantity administered, (
i.e.,
for non-FAMP proposed at § 429.435 and application of MFP proposed at § 429.700). To do so, we need the Part B billing units to be converted to the NCPDP level to match the units on PDE records. We recognize that the Part B data converted to the NCPDP level to match the units in PDE records are not needed for the calculation of the payment amount under section 1847A (b)(4) of the Act as we weight across the Part B and Part D amounts of the selected drug using the 30-day equivalent supply (as set forth in § 429.430), if applicable; however, we still conduct the NCPDP unit conversion to calculate the PDE equivalent number of units here for later use in the non-FAMP ceiling and MFP calculations.
We propose at § 429.425(a)(4) how we would calculate the payment amount under section 1847A(b)(4) of the Act for a 30-day equivalent supply of the selected drug.
We propose at § 429.425(b), that the sequestration payment adjustment is not applied to the amount described under section 1847A(b)(4) of the Act. The amount described under section 1847A(b)(4) of the Act is not adjusted to account for sequestration. Therefore, we do not intend to apply sequestration to the payment amount under section 1847A(b)(4) of the Act as part of the methodology to calculate the payment amount under section 1847A(b)(4) of the Act.
7. Determination of the Combined Part B and Part D Amount (§ 429.430)
Section 1194(c)(1)(B) of the Act provides for the calculation of “an amount,” in the singular, under such subparagraph for each selected drug. Section 1194(c)(1)(B)(ii) of the Act then specifies the payment amount under section 1847A(b)(4) of the Act as an amount to be used under section 1194(c)(1)(B) of the Act for selected drugs that are payable under Part B and paid under section 1847A(b)(4) of the Act and separately specifies at section 1194(c)(1)(B)(i) of the Act the sum of the plan-specific enrollment weighted amounts as an amount to be used for under section 1194(c)(1)(B) of the Act for selected drugs covered under Part D. It does not specify that we should select only one of these two different amounts when calculating the amount for under section 1194(c)(1)(B) of the Act for a selected drug is both covered under Part D and payable under Part B.
For such selected drugs, we interpret section 1194(c)(1)(B) of the Act, which states “. . . an amount equal to . . .” when referring to calculating a single amount under such subparagraph for each selected drug, to mean that we should calculate an amount that captures both the payment amount under section 1847A(b)(4) of the Act and the sum of the plan-specific enrollment weighted amounts, where both amounts are available for such drug (that is, where such drug is payable under Part B and is paid according to section 1847A(b)(4) of the Act). Our proposed interpretation gives effect to the entirety of section 1194(c)(1)(B) of the Act for such drugs when both amounts described thereunder are applicable. In contrast, we do not believe section 1194(c)(1)(B) of the Act is best read as permitting the selection and use of just one of the two applicable amounts under such subparagraph. Such subparagraph offers no criteria by which CMS would choose which of the amounts described thereunder to apply for selected for which both amounts are applicable. Moreover, we do not believe section 1194(c)(1) of the Act is best read
( printed page 36280)
as permitting CMS to use only the amount described in section 1194(c)(1)(C) of the Act for such drugs, because both amounts described under section 1194(c)(1)(B) of the Act are applicable to such drugs and CMS can calculate a single amount that accounts for both such amounts.[52]
To calculate the amount specified at section 1194(c)(1)(B) of the Act for such selected drugs, we propose at § 429.430, consistent with the policies for implementation as described in section 60.2.2.3 of the Negotiation Program Guidance, a methodology to calculate a weighted average of the payment amount under section 1847A(b)(4) of the Act, proposed at § 429.425 and discussed at section II.E.6. of this proposed rule, and the sum of the plan-specific enrollment weighted amounts, proposed at § 429.420 and discussed at section II.E.5. of this proposed rule. We propose at § 429.430(a) that this proposed amount be referred to as the combined Part B and Part D amount. The combined Part B and Part D amount would be for all dosage forms and strengths of a selected drug that are payable under Part B and paid according to section 1847A(b)(4) of the Act and covered under Part D as proposed at § 429.410(b)(2)(iii) and discussed at section II.E.3. of this proposed rule. We propose at § 429.430 how the 30-day equivalent supply will be calculated for the combined Part B and Part D amount.
For the utilization weighting proposed at § 429.430(a)(2), we would treat the NDC-11s of drugs payable under Part B and covered under Part D as having two distinct versions and keep those versions separate in the utilization weighting so that the NDC-11s of drugs payable under Part B and covered under Part D contribute separately to the single amount, based on their applicable proportions to the total. We believe this step is necessary to account for differences in the pricing data between Part D and Part B.
8. Determination of the Applicable Average Non-FAMP Amounts and Applicable Percent of the Average Non-FAMP (§ 429.435)
Section 1194(c)(1)(C)(ii) of the Act requires that for initial price applicability year 2027 and subsequent years, that the lower of the average non-FAMP amounts described in sections 1194(c)(1)(C)(ii)(I) and 1194(c)(1)(C)(ii)(II) of the Act is used as the average non-FAMP amount. Section 1194(c)(1)(C)(ii)(I) of the Act describes the first amount as the average non-FAMP for such drug for 2021 (or, in the case that there is not an average non-FAMP available for such drug for 2021, for the first full year following the market entry for such drug), increased by the percentage increase in the CPI-U from September 2021 (or December of such first full year following the market entry), as applicable, to September of the year prior to the year of the selected drug publication date with respect to such initial price applicability year. Section 1194(c)(1)(C)(ii)(II) of the Act describes the second amount as the average non-FAMP for such drug for the year prior to the selected drug publication date with respect to such initial price applicability year. Consistent with the policies for implementation as described in section 60.2.3 of the Negotiation Program Guidance and in accordance with section 1194(c)(1)(C)(ii) of the Act, we propose at § 429.435(a) to use the methodology and comparison described to determine the lower of the two applicable non-FAMP amounts and apply the applicable percent described in section 1194(c)(3) of the Act.
We propose at § 429.435(a)(1)(i) to use the list of NDC-11s of the selected drug (as determined in proposed § 429.100(c) and discussed in section II.B.1. of this proposed rule), to determine which NDC-11s of the selected drug are included in this ceiling calculation.
Consistent with the policies for implementation as described in section 60.2.3 of the Negotiation Program Guidance, we propose at § 429.435(a)(2)(ii) to use the same methodology for calculating the average non-FAMP for the calendar year 3 years prior to the initial price applicability year of the selected drug as used for the calculation proposed at § 429.435(a)(1)(ii) through (iv) for calendar year 2021, noting that the set of NDCs used to calculate the annual average non-FAMP calculation for each may differ.
To directly compare the amount calculated based on the applicable percent of average non-FAMP proposed in § 429.435 to the amount calculated based on the sum of the plan-specific enrollment weighted amounts proposed in § 429.420, the payment amount under section 1847A(b)(4) of the Act § 429.425, and the combined Part B and Part D amount described in proposed § 429.430, as applicable, we propose to base the average non-FAMP calculations on a 30-day equivalent supply. We propose to use the approach set forth in proposed § 429.435(a)(3) to calculate the average non-FAMP for a 30-day equivalent supply because it is necessary for the calculated non-FAMP amounts to account for different units and treatment regimens across dosage forms and strengths.
We propose at § 429.435(a)(4) to determine the average non-FAMP across all NDC-11s of the selected drug by conducting the steps outlined in this section separately for the average non-FAMP in calendar year 2021 (or for the first full year following market entry for such drug if there is not a non-FAMP for such drug or an average non-FAMP cannot be calculated) and for the calendar year that is 3 years prior to the initial price applicability year of the selected drug. Consistent with the policies for implementation as described in section 60.2.3 of the Negotiation Program Guidance, we propose at § 429.435(a)(4) to calculate an average non-FAMP that is comparable to the sum of the plan-specific enrollment weighted amount (as set forth in § 429.420), the payment amount under section 1847A(b)(4) of the Act (as set forth in § 429.425), or the combined Part B and Part D amount (as set forth in § 429.430), as applicable, and determine the total number of NCPDP units per NDC-11 package. We propose at § 429.435(a)(4)(i) and (ii) the calculations to account for non-FAMP unit volume fluctuations that may occur across quarters.
Consistent with policies for implementation as described in section 60.2.3 of the Negotiation Program Guidance and in accordance with section 1194(c)(1)(C)(ii)(I) of the Act, we propose at § 429.435(a)(4)(v) to increase the average non-FAMP per unit for calendar year 2021 (or for the first full year following market entry for such drug if there is not a non-FAMP for such drug or an average non-FAMP cannot be calculated), which would be calculated in § 429.435(a)(4)(iv) by the percentage increase in CPI-U from September 2021 to September of the calendar year that is 3 years prior to the initial price applicability year of the selected drug.
Consistent with the policies for implementation as described in section 60.2.3 of the Negotiation Program Guidance and in accordance with section 1194(c)(3) of the Act, we propose at § 429.435(a)(4)(vi) to apply the applicable percent to the average non-FAMP associated with the monopoly type for the selected drug that is described in section 1194(c)(3)(A) through (C) of the Act. The applicable
( printed page 36281)
percent is determined based on the initial approval date, as set forth in § 429.125(a)(1)(i), of the selected drug and the initial price applicability year for which the drug is selected for negotiation. We note that applying the applicable percent at proposed § 429.435(a)(4)(vi) results in the same ultimate amount calculated at proposed § 429.435(a)(4)(viii) as it would if we were to apply the applicable percent to the average non-FAMP per 30-day equivalent supply for the selected drug at proposed § 429.435(a)(4)(viii). Consistent with section 1194(c)(3)(A) of the Act, short monopoly drugs and vaccines with respect to a selected drug (other than an extended-monopoly drug and long-monopoly drug), will be 75 percent. Consistent with section 1194(c)(3)(B) of the Act, the applicable percent with respect to an extended-monopoly drug will be 65 percent. Consistent with section 1194(c)(4)(C) of the Act, the applicable percent with respect to long-monopoly drug will be 40 percent.
We propose at § 429.435(a)(4)(vii) and (viii) the steps that we would take for each NDC-11 to calculate the average non-FAMP per 30-day equivalent supply. We propose at § 429.435(a)(4)(ix) the method to compare the amounts calculated for the two separate applicable percent of the average non-FAMP amounts and to determine the lower amount.
9. Temporary Floor for Small Biotech Drugs (§ 429.440)
a. Definitions
In proposed § 429.440, we propose to codify the following definitions applicable to § 429.440 based on policies detailed in this proposed rule.
(1) “Part B 2021 Manufacturer”
We propose to define “Part B 2021 Manufacturer” to mean the NDA holder or the BLA holder for the qualifying single source drug on December 31, 2021.
(2) “Part D 2021 Manufacturer”
We propose to define “Part D 2021 Manufacturer” as the entity that either (1) had a Medicare Coverage Gap Discount Program (CGDP) Agreement under section 1860D-14A of the Act in effect for the qualifying single source drug on December 31, 2021, or (2) had an arrangement whereby the manufacturer's labeler codes were listed on another manufacturer's Medicare CGDP Agreement, consistent with section 1860D-14A of the Act, in effect on December 31, 2021.
(3) “Part B 2021 Manufacturer and Its Controlled Group”
We propose to define “Part B 2021 Manufacturer and its controlled group” as comprising all persons that, as of December 31, 2021, were treated as a single employer under subsection (a) or (b) of section 52 of the Internal Revenue Code of 1986 with the Part B 2021 Manufacturer.
(4) “Part D 2021 Manufacturer and Its Controlled Group”.
We propose to define “Part D 2021 Manufacturer and its controlled group” as comprising all persons that, as of December 31, 2021, were treated as a single employer under subsection (a) or (b) of section 52 of the Internal Revenue Code of 1986 with the Part D 2021 Manufacturer and had a CGDP Agreement in effect on December 31, 2021.
b. Implementation of the Temporary Floor for Small Biotech Drugs
Section 1192(d)(2) of the Act established the Small Biotech Exception (SBE), which states that a negotiation eligible drug shall not include, with respect to initial price applicability years 2026, 2027, and 2028, a qualifying single source drug that meets either the criteria at section 1192(d)(2)(A)(i) or section 1192(d)(2)(A)(ii) of the Act. With respect to initial price applicability years 2026 through 2028, CMS implemented the requirements set forth in section 1192(d)(2) of the Act for a qualifying single source drug to be determined a Small Biotech Drug and therefore eligible for the SBE through guidance, including for example, section 30.2.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
Section 1194(d) of the Act requires that for a selected drug that is a qualifying single source drug that meets the requirements set forth in section 1192(d)(2) of the Act to be determined a Small Biotech Drug, and with respect to which the first initial price applicability year of the price applicability period with respect to such drug is 2029 or 2030, CMS will not offer or agree to a counteroffer for an MFP that is lower than the Temporary Floor for Small Biotech Drugs. To fulfill the statutory obligation at section 1194(f)(4)(B) of the Act, which states that reference to the first initial price applicability year of the price applicability period with respect to such drug shall be treated as the first initial price applicability year of such period for which the maximum fair price established pursuant to renegotiation applies, drugs selected for renegotiation also would be eligible to be considered for the Temporary Floor for Small Biotech Drugs described in section 1194(d). Section 1194(d) of the Act states that the Temporary Floor for Small Biotech Drugs shall be equal to 66 percent of the average non-FAMP for such drug for 2021 (or, in the case that there is not an average non-FAMP available for such drug for 2021, for the first full year following the market entry for such drug), increased by the percentage increase in the CPI-U from September 2021 (or December of such first full year following the market entry), as applicable, to September of the year prior to the year of the selected drug publication date with respect to such initial price applicability year.
With respect to a drug selected for negotiation for initial price applicability year 2029 and 2030, or a drug selected for renegotiation for initial price applicability year 2029 or 2030, for which the Primary Manufacturer submits information in accordance with § 429.440(b)(1), we propose at § 429.440(b) to establish the Temporary Floor for Small Biotech Drugs for selected drugs that are determined to be a Small Biotech Drug based on the criteria that were used and implemented with respect to the SBE for initial price applicability year 2028, as proposed in § 429.440(b)(2). We will not make an offer or agree to a counteroffer for an MFP (as proposed in § 429.500(b) and described in section II.F.1. of this proposed rule) that is lower than the Temporary Floor for Small Biotech Drugs, which shall be equal to the amount specified in proposed § 429.440(b)(3). A determination by CMS that a given selected drug is eligible for the Temporary Floor for Small Biotech Drugs for initial price applicability years 2029 and 2030 is not based on whether or not CMS previously determined that the qualifying single source drug was eligible or not eligible for the SBE for initial price applicability years 2026, 2027, or 2028.
We propose at § 429.440(b)(1) to establish a process whereby a Primary Manufacturer that would like its selected drug to be considered eligible for the Temporary Floor for Small Biotech Drugs must submit information to allow CMS to determine whether its selected drug meets the requirements of a Small Biotech Drug. This submission is a prerequisite for the Temporary Floor for Small Biotech Drugs to apply to a selected drug. We are proposing at § 429.440(b)(2) to establish requirements for determining whether a selected drug is a Small Biotech Drug that are
( printed page 36282)
consistent with the eligibility requirements for the SBE for initial price applicability year 2028, which were implemented through section 30.2.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028. If a selected drug is determined to be a Small Biotech Drug, CMS would then calculate the Temporary Floor for Small Biotech Drugs, as proposed at § 429.440(b)(3). We propose at § 429.440(b)(4) to establish a stepwise approach to adjust the ceiling for a selected drug in the event that the ceiling identified in § 429.410(b) (or § 429.620(b) as applicable) is below the Temporary Floor for Small Biotech Drugs, if applicable.
As discussed in further detail in section IV. of this proposed rule, for a Primary Manufacturer to be considered for the Temporary Floor for Small Biotech Drugs, we propose revisions to a currently approved information collection request, titled the Negotiation Program Drug Price Negotiation for Initial Price Applicability Year 20XX under Section 11001 and 11002 of the Inflation Reduction Act Information Collection Request (ICR) (CMS-10849, OMB 0938-1452) (hereinafter, the “Drug Price Negotiation ICR”), for a 60-day public comment period concurrently with this proposed rule. A form and manner for submitting an application for the Temporary Floor for Small Biotech Drugs, consistent with proposed § 429.440(b)(1), would be specified in the ICR for initial price applicability year 2029 and initial price applicability year 2030. Information submitted in an application for the Temporary Floor for Small Biotech Drugs that is a trade secret or confidential commercial or financial information will be protected from disclosure if the information meets the requirements set forth under Exemptions 3 and/or 4 of the Freedom of Information Act (FOIA) (5 U.S.C. 552(b)(3), (4)).
In accordance with section 1194(d) of the Act, to determine eligibility for the Temporary Floor for Small Biotech Drugs, and proposed at § 429.440(b)(2), we would determine whether the selected drug meets the criteria set forth in section 1192(d)(2) [53]
of the Act. Section 1192(d)(2)(A)(i) of the Act establishes that for a selected drug that is covered under Part D to be considered a Small Biotech Drug, the Total Expenditures under Part D during 2021 for such drug must be: equal to or less than 1 percent of the Total Expenditures under Part D for all covered Part D drugs during 2021; and equal to or greater than 80 percent of the Total Expenditures under Part D for all covered Part D drugs for which the manufacturer had a Coverage Gap Discount Program Agreement in effect during 2021. Section 1192(d)(2)(A)(ii) of the Act establishes that for a selected drug that is payable under Part B to be considered a Small Biotech Drug, the Total Expenditures under Part B for such drug during 2021 must be: equal to or less than 1 percent of the Total Expenditures under Part B for all qualifying single source drugs for which payment may be made under Part B during 2021; and equal to or greater than 80 percent of the Total Expenditures under Part B for all qualifying single source drugs of the manufacturer for which payment may be made under Part B during 2021. A selected drug that meets either the criteria set forth in section 1192(d)(2)(A)(i) of the Act or the criteria set forth in section 1192(d)(2)(A)(ii) of the Act will qualify as a Small Biotech Drug. Section 1192(d)(2)(B)(i) of the Act establishes the aggregation rule, which establishes that all persons treated as a single employer under subsection (a) or (b) of section 52 of the Internal Revenue Code of 1986 shall be treated as one manufacturer when determining whether a selected drug is a Small Biotech Drug. Additionally, section 1192(d)(2)(B)(ii) of the Act establishes that a selected drug shall not be considered a Small Biotech Drug if the manufacturer of such drug is acquired after 2021 by another manufacturer that does not meet the definition of a specified manufacturer under section 1860D-14C(g)(4)(B)(ii) of the Act, effective at the beginning of the plan year immediately following such acquisition or, in the case of an acquisition before 2025, effective January 1, 2025. With respect to initial price applicability years 2026 through 2028, CMS implemented the requirements set forth in sections 1192(d)(2)(A)(i), 1192(d)(2)(A)(ii), 1192(d)(2)(B)(i), and 1192(d)(2)(B)(ii) of the Act through guidance, including, for example, section 30.2.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability years 2029 and 2030, and consistent with the policies for implementation of the SBE through section 30.2.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028, we propose at § 429.440(b)(2)(i) to establish the Part D Track, which outlines the criteria for determining whether a selected drug covered under Part D qualifies as a Small Biotech Drug in accordance with section 1192(d)(2)(A)(i) of the Act. In our assessment, we would consider Total Expenditures under Part D for all covered Part D drugs during 2021, Total Expenditures under Part D for the selected drug during 2021, and Total Expenditures under Part D during 2021 for all covered Part D drugs for which the Part D 2021 Manufacturer (as defined in proposed § 429.440(a)(2)) and its controlled group (as defined in proposed § 429.440(a)(4)) had a CGDP Agreement in effect on December 31, 2021, as calculated using the methodology proposed in § 429.120(a) and described section in II.B.5.a. of this proposed rule, except the date of service as described in § 429.120(a)(1) is during 2021. Specifically, we would consider whether, for dates of service in calendar year 2021, the Total Expenditures during 2021 under Part D for the selected drug were: (1) equal to or less than 1 percent of the Total Expenditures under Part D for all covered Part D drugs during 2021; and (2) equal to or greater than 80 percent of the Total Expenditures under Part D for all covered Part D drugs during 2021 for which the Part D 2021 Manufacturer and its controlled group had a CGDP Agreement in effect on December 31, 2021. For the purpose of performing the calculations described at proposed § 429.440(b)(2)(ii), CMS would identify the NDC-11s of the selected drug determined under § 429.100(d) with respect to the initial price applicability year for which the Primary Manufacturer of the selected drug applied for the Temporary Floor for Small Biotech Drugs in accordance with proposed § 429.440(b)(1). We would identify the drugs covered under Part D for which the Part D 2021 Manufacturer and its controlled group had a CGDP Agreement in effect on December 31, 2021 as the NDC-11s corresponding to the labeler codes of the Part D 2021 Manufacturer and its controlled group.
For the purposes of determining whether a selected drug qualifies as a Small Biotech Drug via the Part D Track, we need to collect information to
( printed page 36283)
accurately identify the Part D 2021 Manufacturer, which is the entity that had the CGDP Agreement under section 1860D-14A of the Act in effect for the selected drug on December 31, 2021. In addition, in accordance with section 1192(d)(2)(B)(i) of the Act, we propose at § 429.440(a)(4) that the Part D 2021 Manufacturer and its controlled group comprise all persons that, as of December 31, 2021, were treated as a single employer with the Part D 2021 Manufacturer and had a CGDP Agreement in effect on December 31, 2021. However, CMS does not have information about which entities were treated as a single employer with the Part D 2021 Manufacturer under the applicable IRC provisions and the Treasury regulations thereunder. Therefore, we propose that a Primary Manufacturer that seeks the Temporary Floor for Small Biotech Drugs for its selected drug covered under Part D would be required to submit information to CMS about the Part D 2021 Manufacturer and its controlled group to be considered for the Part D Track.
With respect to initial price applicability years 2029 and 2030, and consistent with the policies for implementation of the SBE, we propose at § 429.440(b)(2)(ii) to establish the Part B Track, which outlines the criteria for determining whether a selected drug payable under Part B qualifies as a Small Biotech Drug in accordance with section 1192(d)(2)(A)(ii) of the Act. In our assessment, we would consider Total Expenditures under Part B during 2021 for all qualifying single source drugs, Total Expenditures under Part B during 2021 for the selected drug, and Total Expenditures under Part B in 2021 for all qualifying single source drugs of the Part B 2021 Manufacturer (as defined in proposed § 429.440(a)(1)) and its controlled group (as defined in proposed § 429.440(a)(3)), calculated using the methodology proposed in § 429.120(b) and described in II.B.5.b. of this proposed rule, except the date of service as described in § 429.120(b)(1)(i) and (b)(2)(i) is during 2021. We would consider whether, for Part B data, as defined in § 429.20, with dates of service during 2021, the Total Expenditures under Part B during 2021 for the selected drug were: (1) equal to or less than 1 percent of the Total Expenditures under Part B for all qualifying single source drugs payable under Part B during 2021; and (2) equal to at least 80 percent of the Total Expenditures under Part B during 2021 for all qualifying single source drugs of the Part B 2021 Manufacturer and its controlled group for which payment may be made under Part B. For the purpose of performing the calculations described at proposed § 429.440(b)(2)(ii), CMS would identify the NDC-11s of the selected drug determined under § 429.100(d) with respect to the initial price applicability year for which the Primary Manufacturer of the selected drug applied for the Temporary Floor for Small Biotech Drugs in accordance with proposed § 429.440(b)(1). CMS would identify the NDC-11s of other applicable qualifying single source drugs using the policies for identifying qualifying single source drugs proposed in § 429.125. Accordingly, we would identify the qualifying single source drug(s) payable under Part B for the Part B 2021 Manufacturer and its controlled group using the NDC-11(s) associated with the qualifying single source drug(s) that correspond to the NDA(s) and/or BLA(s) held by the Part B 2021 Manufacturer or any member of its controlled group on December 31, 2021. For the purposes of determining whether a selected drug qualifies as a Small Biotech Drug via the Part B Track, we need to collect information to accurately identify the Part B 2021 Manufacturer, which is the entity that is the NDA(s) holder or BLA(s) holder for the selected drug on December 31, 2021. In addition, in accordance with section 1192(d)(2)(B)(i) of the Act, we propose at § 429.440(a)(3) that the Part B 2021 Manufacturer and its controlled group comprise all persons that, as of December 31, 2021, were treated as a single employer with the Part B 2021 Manufacturer. However, CMS does not have information about which entities were treated as a single employer with the Part B 2021 Manufacturer under the applicable IRC provisions and the Treasury regulations thereunder. Therefore, we propose that a Primary Manufacturer that seeks the Temporary Floor for Small Biotech Drugs for its selected drug covered under Part B would be required to submit information to CMS about the Part B 2021 Manufacturer and its controlled group to be considered for the Part B Track.
In addition, in accordance with section 1192(d)(2)(B)(i) of the Act, we propose at § 429.440(b)(2)(iii) to preclude a selected drug from being eligible for the Temporary Floor for Small Biotech Drugs if the Primary Manufacturer of such drug is acquired after 2021 by another manufacturer that does not meet the definition of a specified manufacturer under section 1860D-14C(g)(4)(B)(ii) of the Act, effective at the beginning of the plan year immediately following such acquisition or, in the case of an acquisition before 2025, effective January 1, 2025.[54]
For purposes of implementing this limitation, we would use the determinations of the Manufacturer Discount Program as to whether the acquiring entity met the definition of specified manufacturer in the applicable period. We would consider an acquiring entity to have met the Manufacturer Discount Program definition of specified manufacturer for purposes of this limitation if the acquiring entity is identified by CMS under the Manufacturer Discount Program as either a specified manufacturer under section 1860D-14C(g)(4)(B)(ii) of the Act or a specified small manufacturer under section 1860D-14C(g)(4)(C)(ii) of the Act.[55]
For an acquisition of a Primary Manufacturer to be relevant to the limitation, and therefore to potentially preclude a selected drug from being considered a Small Biotech Drug, we propose that the transaction must occur after 2021 and must involve the acquisition of the Primary Manufacturer after it held the NDA(s) or BLA(s) for the drug.
In accordance with section 1194(d) of the Act, we propose at § 429.440(b)(3) to calculate the Temporary Floor for Small Biotech Drugs for each Small Biotech Drug as 66 percent of the average non-FAMP in calendar year 2021, calculated using the methodology set forth in § 429.435, increased by the percentage increase in the CPI-U from September 2021 to September of the year prior to the selected drug publication date for which the drug is selected for negotiation or, as applicable, renegotiation. If a selected drug did not have a non-FAMP for 2021, we would use the average non-FAMP for the first full year following the market entry, increased by the percentage increase in the CPI-U from December of the first full year following market entry to September of the year prior to the selected drug publication date for which
( printed page 36284)
the drug is selected for negotiation or, as applicable, renegotiation.
In accordance with section 1194(b)(2)(F)(ii) of the Act and as proposed in § 429.410(a), we would not offer or agree to a counteroffer for an MFP that is below the Temporary Floor for Small Biotech Drugs. However, we note that there is a possibility under the statute that the ceiling calculated as described in proposed § 429.410(b) (or § 429.620(b), as applicable) is below the Temporary Floor for Small Biotech Drugs. The statute does not expressly address how to apply section 1194(b)(2)(F)(ii) of the Act in such cases, though such a scenario is possible under the statute—for example, if a selected drug is eligible for the Temporary Floor for Small Biotech Drugs in initial price applicability year 2030 and is an extended-monopoly drug, the applicable percent of average non-FAMP for 2021 used in the ceiling calculation for an extended-monopoly drug under proposed § 429.435(a)(4)(vii)(A)(2) would be less than that of the Temporary Floor for Small Biotech Drugs under proposed § 429.440(b)(3) (65 percent and 66 percent, respectively).
Additionally, as proposed in § 429.620(b) and further described in section II.G.5. of this proposed rule, for the purpose of calculating the ceiling for renegotiation, we propose to determine the ceiling applicable to renegotiation using the ceiling amounts determined with respect to the selected drug's original negotiation, with limited updates, including to account for the statutory directive to update the applicable percent for certain renegotiation-eligible drugs. However, for drugs selected for renegotiation that are eligible for the Temporary Floor for Small Biotech Drugs, the ceiling determined under § 429.620(b) may be lower than the Temporary Floor for Small Biotech Drugs.
To address such a scenario, and to ensure the negotiation process gives effect to the Temporary Floor for Small Biotech Drugs provision at section 1194(d) of the Act that establishes a specific limitation to the general rules for MFP negotiations, we are proposing at § 429.440(b)(4) a stepwise approach to calculate an adjusted ceiling that would apply to the negotiation, or renegotiation, process for selected drugs that qualify for the Temporary Floor for Small Biotech Drugs. We would first determine if an adjusted ceiling is needed by calculating the unadjusted ceiling as described at proposed § 429.410(b) or the renegotiated unadjusted ceiling as described in § 429.620(b) (as applicable), as well as the Temporary Floor for Small Biotech drugs as described at proposed § 429.440(b)(3), and then would determine if the unadjusted ceiling is below the Temporary Floor for Small Biotech Drugs. If the unadjusted ceiling is greater than or equal to the Temporary Floor for Small Biotech Drugs, then this ceiling would apply, and we would not offer or accept a counteroffer for an MFP that is below the Temporary Floor for Small Biotech Drugs or greater than the ceiling for the selected drug. If, however, such ceiling is below the Temporary Floor for Small Biotech Drugs, we would calculate an adjusted ceiling, as described in proposed § 429.440(b)(4)(i) or (ii), as applicable. To calculate an adjusted ceiling, we propose in § 429.440(b)(4)(i) that, for purposes of determining the potential ceiling amount described under section 1194(c)(1)(C) of the Act, rather than selecting the lower of the average non-FAMP available for such drug in 2021 (or the first full year following market entry for such drug if there is not a non-FAMP for such drug or an average non-FAMP cannot be calculated), as described in section 1194(c)(1)(C)(i) or section 1194(c)(1)(C)(ii)(I) of the Act and proposed § 429.435(a)(1) or § 429.620(b) (as applicable), or the average non-FAMP for such drug for the year prior to the selected drug publication date, as described in section 1194(c)(1)(C)(ii)(II) of the Act and proposed § 429.435(a)(2) or § 429.620(b) (as applicable), to calculate the lower amount of the applicable percent of the average non-FAMP, as described in proposed § 429.410(b) (or § 429.620(b) for both non-FAMPs as applicable), we would only consider the average non-FAMP for such drug for the prior year to the selected drug publication date (that is, the amount described in section 1194(c)(1)(C)(ii)(II) of the Act). Therefore, the adjusted ceiling proposed in § 429.440(b)(4)(i) would determine the ceiling as described in § 429.410(b) (or § 429.620(b) as applicable) except that the lower amount of the applicable percent of the average non-FAMP, as proposed in § 429.435 (or § 429.620(b) as applicable), would consider only the average non-FAMP for such drug for the prior year to the selected drug publication date, as described in § 429.435(a)(2) (or § 429.620(b), as applicable). We believe that removing the average non-FAMP available for such drug in 2021 from the calculation of the lower amount of the applicable percent of the average non-FAMP allows for the implementation of the Temporary Floor for Small Biotech Drugs while preserving the ceiling calculation, giving effect to both provisions, and is the most appropriate and narrowly tailored way to reconcile cases where the Temporary Floor for Small Biotech Drugs exceeds the ceiling for a selected drug, as the average non-FAMP available for such drug in 2021 may be in direct tension when calculating the ceiling and the Temporary Floor for Small Biotech Drugs, due the applicable percent being applied in contradictory ways in the ceiling and the Temporary Floor for Small Biotech Drugs. If this adjusted ceiling is greater than or equal to the Temporary Floor for Small Biotech Drugs, then such adjusted ceiling would be used as the ceiling for the negotiation between CMS and the Primary Manufacturer, and we would not offer or agree to a counteroffer for an MFP that is below the Temporary Floor for Small Biotech Drugs or greater than this adjusted ceiling for the selected drug.
If this adjusted ceiling is still below the Temporary Floor for Small Biotech Drugs, then, as described in proposed § 429.440(b)(4)(ii), we would raise the ceiling to be equal to the Temporary Floor for Small Biotech Drugs. We believe that raising the ceiling to be equal to the Temporary Floor for Small Biotech Drugs appropriately balances the statutory requirements to establish a ceiling for a selected drug while ensuring the negotiation process also gives full effect to the Temporary Floor for Small Biotech Drugs provision at section 1194(d) of the Act. We note that the Temporary Floor for Small Biotech Drugs provision applies only with respect to negotiations and renegotiations that occur for initial price applicability years 2029 and 2030 for eligible drugs, and it would not apply to any subsequent renegotiations of drugs selected for renegotiation with respect to an initial price applicability year after initial price applicability year 2030. We considered disregarding the ceiling entirely, but we do not believe this approach would align with the statutory requirements for both a ceiling and a Temporary Floor for Small Biotech Drugs. We also considered using the amount calculated in section 1194(c)(1)(B) of the Act (as proposed in § 429.410(b)(1) or § 429.620(b)) as the sole amount for the ceiling, however we do not believe this approach would align with the statutory requirements to apply an applicable percent in section 1194(c)(1)(C) of the Act, and would result in a ceiling that would not distinguish between a short-monopoly drug, a long-monopoly drug, and an extended-monopoly drug (beginning in
( printed page 36285)
initial price applicability year 2030). In accordance with section 1194(b)(2)(F) of the Act, because the ceiling and the Temporary Floor for Small Biotech Drugs for a selected drug would equal the same price, we would only offer or agree to a counteroffer for an MFP that is equal to such price. We believe that this stepwise approach addresses circumstances in which the ceiling is below the Temporary Floor for Small Biotech Drugs while maintaining the integrity of the negotiation process and implementing the statutory responsibility to not offer or accept a counteroffer of an MFP that is above the ceiling or below the Temporary Floor for Small Biotech Drugs.
As proposed in § 429.440(b)(5), if a Primary Manufacturer submits an application for the Temporary Floor for Small Biotech Drugs, we would provide a notice in writing to the Primary Manufacturer, alongside the calculation information provided as set forth in § 429.445(a), which would include: a determination of whether the selected drug is a Small Biotech Drug; and, if the selected drug is eligible for the Temporary Floor for Small Biotech Drugs, the calculation of the Temporary Floor for Small Biotech Drugs as proposed in § 429.440(b)(3) (or adjusted ceiling, if applicable, as proposed in § 429.440(b)(4)). We propose in § 429.440(b)(6) to allow a Primary Manufacturer that believes in good faith that CMS has made an error in calculations pertaining to the calculation of the Temporary Floor for Small Biotech Drugs, as proposed in § 429.440(b)(3), or the adjusted ceiling, if applicable, calculated for a drug that receives the Temporary Floor for Small Biotech Drugs, as proposed in § 429.440(b)(4), to submit a Suggestion of Error as proposed in § 429.445(c).
10. Calculation Information and Suggestion of Error (§ 429.445)
In implementing the statutory provisions of the Negotiation Program, CMS is required to execute a number of multi-step calculations. Section 1194(b)(2)(F)(i) of the Act requires that, in negotiating the MFP of a selected drug, including during renegotiation, CMS would not make an offer or agree to a counteroffer for an MFP that exceeds the ceiling specified in section 1194(c) of the Act. Section 1196(a) of the Act describes certain of CMS' administrative duties, including section 1196(a)(2) of the Act, which requires CMS to establish procedures to compute and apply the MFP across dosage forms and strengths of a selected drug and not based on the specific formulation or package size or package type of such drug. Section 1194(d) of the Act requires that for a selected drug that is a qualifying single source drug that meets the requirements set forth in section 1192(d)(2) of the Act to be determined a Small Biotech Drug, and with respect to which the first initial price applicability year of the price applicability period with respect to such drug is 2029 or 2030, CMS will not offer or agree to a counteroffer for an MFP that is lower than the Temporary Floor for Small Biotech Drugs. With respect to initial price applicability years 2026 through 2028, we implemented a suggestion of error process for Primary Manufacturers if they believe in good faith that CMS has made an error in calculating the ceiling or in applying the MFP across dosage forms and strengths of a selected drug, including, for example, in section 40.5 of the Negotiation Program Guidance with respect to initial price applicability year 2028. With respect to initial price applicability year 2029 and subsequent years, consistent with the policies for implementation as described in section 40.5 of the Negotiation Program Guidance, as revised based on the proposed modifications discussed in this section, we propose in § 429.445 to provide a Primary Manufacturer the opportunity to submit a suggestion of error if they believe in good faith that CMS has made an error in the calculation of the ceiling or the computation of how CMS intends to apply a single MFP across dosage forms and strengths of the selected drug.
We propose at § 429.445(a) to provide a Primary Manufacturer with the following information, as applicable: information on CMS' calculation of the ceiling, as described in §§ 429.410 through 429.435; the computation of how we will apply a single MFP across dosage forms and strengths of the selected drug, as described in § 429.700(b) and (c); and information on CMS' calculations of the Temporary Floor for Small Biotech Drugs (and adjusted ceiling, as applicable), as described in § 429.440(b)(3) and (b)(4). The information on CMS' calculations of the ceiling would include each intermediate step in the calculation of the section 1847A(b)(4) of the Act payment amount as described in § 429.425, and the non-FAMP amounts as described in § 429.435. The plan-specific enrollment weighted amount as described in § 429.420 would also be included in the information on CMS' calculations of the ceiling but the intermediate steps that make up the plan-specific weighted amount would not, as the plan sponsor's reported Part D PDE data is proprietary data of the plan sponsors and is protected from disclosure, as described in the confidentiality policy proposed in § 429.300(b). The computation of how we will apply a single MFP across dosage forms and strengths of the selected drug would include each intermediate step in our calculation for all applicable NDCs associated with a selected drug, as described in § 429.700 to demonstrate how we will apply a single MFP across dosage forms and strengths. If a selected drug is eligible for the Temporary Floor for Small Biotech Drugs, the information on the calculations of the Temporary Floor for Small Biotech Drugs would include each intermediate step in the calculation of the Temporary Floor for Small Biotech Drugs as described in § 429.440(b)(3). We would also include the information on CMS' calculation of the ceiling, as described previously, and information on CMS' calculation of the adjusted ceiling, if applicable. The information on CMS' calculations of the alternative ceiling will include a modified version of the ceiling calculations, based on whether the selected drug's adjusted ceiling is calculated using proposed § 429.440(b)(4)(i) or (ii).
We propose in § 429.445(b)(1) to provide the information on CMS' calculation of the ceiling, the computation of how we will apply a single MFP across dosage forms and strengths of the selected drug, and, as applicable, information on CMS' calculations of the Temporary Floor for Small Biotech Drugs (and adjusted ceiling, as applicable) following the Primary Manufacturer's submission of the section 1194(e)(1) data described in proposed §§ 429.100(d), 429.405(a), 429.440(b)(1), and 429.505(b)(2) and following the Primary Manufacturer's submission of the section 1194(e)(1) data described in proposed § 429.615(b)(1). Additionally, in proposed § 429.445(b)(2), we would also provide the computation of how we will apply a single MFP across dosage forms and strengths of the selected drug following the Primary Manufacturer's submission of any updates to the list of NDC-11s, as proposed in § 429.100(e) and following the determination that an NDC with insufficient data, as described in section II.H.1. of this proposed rule and proposed in § 429.700(c)(3) has sufficient data, as proposed in § 429.700(c)(4)(i)(B)(2).
Further, we propose in § 429.445(c) that Primary Manufacturers would have 10 days to submit a suggestion of error with respect to these calculations if they believe that CMS has made a calculation
( printed page 36286)
error. A calculation error is any error related to the mathematical equations that make up the calculation of the ceiling, the computation of how CMS will apply a single MFP across dosage forms and strengths of the selected drug, or the calculations of the Temporary Floor for Small Biotech Drugs (and adjusted ceiling, if applicable) for a selected drug eligible for the Temporary Floor for Small Biotech Drugs. This includes errors in the sums, products, and quotients of values in the mathematical equations. In contrast, objections to policies and methodologies for implementing the relevant calculations would not be considered a relevant suggestion of error.
Under the applicable guidance for initial price applicability years 2027 and 2028, Primary Manufacturers have 21 days to submit a suggestion of error. We propose to change the length of time that a Primary Manufacturer has to submit a suggestion of error with respect to the information provided under proposed § 429.445(a)(1) and (a)(3) in order to ensure we can meet deadlines for the negotiation process established in statute. As described in section II.C.1. of this proposed rule, in accordance with section 1194(b)(2)(A) of the Act, the Primary Manufacturer of a selected drug must submit, no later than March 1 of the year of the selected drug publication date, with respect to the selected drug, certain information, including non-FAMP data and other information required to carry out the negotiation process, including the information specified at section 1194(e)(1) of the Act. In accordance with section 1194(b)(2)(B) of the Act, and as proposed at § 429.520(a), CMS would provide the Primary Manufacturer with the written initial offer no later than June 1 following the selected drug publication date. Therefore, CMS has only 3 months to process and analyze Primary Manufacturer data that the agency must consider when preparing the written initial offer. Our experience implementing the Negotiation Program to date has demonstrated that a suggestion of error period of 21 days (or more) impedes our ability to perform the analysis necessary to meet the June 1st statutory deadline. In particular, we have found that additional time is needed beyond March 1st to engage with Primary Manufacturers regarding their required data submissions to ensure that they have submitted the data necessary for us to perform the required calculations and that such information is presented with sufficient clarity for us to be able to use it, in accordance with their obligations under the Negotiation Program Agreement. We cannot provide the ceiling calculation information described in proposed § 429.410 through § 429.435, until we have engaged in this process. Reducing the time for Primary Manufacturers to submit a suggestion of error to 10 days will allow for a timely review of Primary Manufacturer submissions of the section 1194(e)(1) factors, the opportunity for Primary Manufacturers to supplement and clarify submissions in accordance with agency requests in accordance with proposed § 429.900(b), and allow for the delivery of the information to the Primary Manufacturer and the suggestion of error window to be complete prior to the June 1st statutory deadline for written initial offers.
Additionally, we propose to change the length of time that a Primary Manufacturer has to submit a suggestion of error with respect to the information provided under proposed § 429.445(a)(2) in order to incorporate Primary Manufacturer feedback to the extent practicable and as appropriate, into MFP application calculations prior to the MFP going into effect. As described in section II.B.1. of this proposed rule, a Primary Manufacturer of a selected drug has an ongoing obligation to report, at least 30 calendar days prior to the change taking effect, any changes to the list of NDC-11s provided in proposed § 429.100(d) to ensure the list of NDC-11s of the selected drug remains complete and accurate. Our experience implementing the Negotiation Program to date has demonstrated that a suggestion of error period of 21 days does not leave the agency sufficient time to consider Primary Manufacturer feedback prior to implementation of revised MFPs and MFPs for new NDCs. Reducing the time for Primary Manufacturers to submit a suggestion of error to 10 days will allow for CMS to consider, and incorporate, to the extent practicable and as appropriate, Primary Manufacturer feedback prior to implementation of revised MFPs and MFPs for new NDCs.
As further specified in proposed § 429.445(c), the suggestion of error process would not affect a Primary Manufacturer's obligation to comply with Negotiation Program requirements and would not alter or change any timelines or requirements of the Negotiation Program.
A Primary Manufacturer must submit any suggestion of error on the calculation of the ceiling, the computation of how CMS would apply a single MFP across dosage forms and strengths, and information on CMS' calculation of the Temporary Floor for Small Biotech Drugs (and adjusted ceiling, as applicable) of the selected drug in a form and manner specified by CMS, as proposed in § 429.445(d).
We solicit comments on our proposed approach for the suggestion of error process. In addition, we solicit comment on whether the regulatory text should expressly identify each of the calculations for which this suggestion of error process would be available.
F. Negotiation Process (§§ 429.500 through 429.535)
1. General Rule (§ 429.500)
Section 1194 of the Act describes the process that CMS must follow when conducting negotiation and renegotiation. Specifically, section 1194(b)(2) of the Act directs CMS to use a consistent methodology and process for negotiation that aims to achieve the lowest MFP for each selected drug. Further, section 1194(e) of the Act requires CMS to consider section 1194(e)(1) factors and section 1194(e)(2) factors as the basis for determining offers and counteroffers. CMS proposes to codify these provisions at § 429.500(a). We also propose in § 429.500(b) that to formalize an agreed-upon MFP, CMS and the Primary Manufacturer must sign an Addendum to the Negotiation Program Agreement, as described in proposed § 429.200(e) and discussed in section II.C.1. of this proposed rule.
With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, sections 60.3 and 60.4 of the Negotiation Program Guidance with respect to initial price applicability year 2028. With respect to initial price applicability year 2029 and subsequent years, we are proposing in §§ 429.500 through 429.535 to codify the negotiation process consistent with the requirements of the IRA and such prior guidance, with proposed revisions noted in these sections.
2. Negotiation Factors (§ 429.505)
Section 1194(e) of the Act directs CMS, for purposes of negotiating the MFP for a selected drug with the Primary Manufacturer, to consider certain factors, as applicable to the selected drug, as the basis for determining offers and counteroffers, as described at proposed § 429.505(a) and further discussed at proposed § 429.510(e) and (f) and section II.F.3. of this proposed rule. These factors include data submitted by the Primary Manufacturer, as specified in section
( printed page 36287)
1194(e)(1) of the Act and evidence about alternative treatments, as specified in section 1194(e)(2) of the Act. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, with respect to initial price applicability year 2028, section 50 of the Negotiation Program Guidance. With respect to initial price applicability year 2029 and subsequent years, consistent with the policies for implementation as described in section 50 of the Negotiation Program Guidance, proposed § 429.505, as described more fully below, codifies our collection of data related to the factors listed at section 1194(e) of the Act, the process for such data collection, the timing of such data collection, and requirements for the Primary Manufacturer to update submitted data. This section also discusses how we propose to consider evidence from comparative clinical effectiveness research in accordance with section 1194(e)(2) of the Act.
a. Information Related to Section 1194(e)(1) Factors
As noted previously, section 1194(e) of the Act directs CMS, for purposes of negotiating the MFP for a selected drug with the Primary Manufacturer, to consider certain factors, as applicable to the selected drug, as the basis for determining offers and counteroffers, including data submitted by the Primary Manufacturer, as specified in section 1194(e)(1) of the Act. Further, section 1194(b)(2)(A) requires that, not later than March 1 of the year of the selected drug publication date, with respect to the selected drug, the Primary Manufacturer shall submit the information described in section 1193(a)(4) of the Act, including but not limited to, information required to carry out the negotiation or renegotiation process. Accordingly, in accordance with the requirements of its Negotiation Program Agreement (proposed in subpart C and described in section II.C.3. of this proposed rule) and in accordance with sections 1193(a)(4) and 1194(b)(2)(A) of the Act, we propose to codify at § 429.505(b) that the Primary Manufacturer of a selected drug is required to submit, among other information, information required to carry out the Negotiation Program, including but not limited to, information related to the factors listed in section 1194(e)(1) of the Act, inclusive of NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer, by 11:59 p.m. PST on March 1 of the year of the selected drug publication date (described in proposed § 429.100(c) and section II.B.1. of this proposed rule). We propose that such information would be submitted through the Drug Price Negotiation ICR.
Specifically, in accordance with sections 1193(a)(4), 1194(b)(2)(A), and 1194(e)(1) of the Act, we propose at § 429.505(b)(1) to codify that a Primary Manufacturer is required to submit the following information with respect to its selected drug, inclusive of NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer, by 11:59 p.m. PST on March 1 of the year of the selected drug publication date: (1) R&D costs of the Primary Manufacturer for the selected drug and the extent to which the Primary Manufacturer has recouped those costs; (2) current unit costs of production and distribution of the selected drug; (3) prior Federal financial support for novel therapeutic discovery and development with respect to the selected drug; (4) data on pending and approved patent applications, exclusivities recognized by the FDA, and applications and approvals under section 505(c) of the FD&C Act or section 351(a) of the PHS Act for the selected drug; and (5) market data and revenue and sales volume data for the selected drug in the United States.
When determining what information we require to carry out the statutory obligation to consider the section 1194(e)(1) factors, we considered the operational and financial burden to the Primary Manufacturer alongside the importance of collecting sufficient information to inform determination of offers and counteroffers, including development of the initial offer. Specifically for the section 1194(e)(1) factors, we do not believe that we are able to obtain this information from sources other than the Primary Manufacturer of the selected drug per statute regardless of whether alternative sources for this information may exist, whether publicly available or available within CMS. Likewise, in accordance with section 1194(e)(1) of the Act, the Primary Manufacturer will be required to submit information for the section 1194(e)(1) factors for renegotiation, as proposed in § 429.615(b)(1) and discussed further in II.G.4 of this proposed rule.
In accordance with a Primary Manufacturer's responsibility under section 1193(a)(5) of the Act and under the Negotiation Program Agreement (set forth in proposed § 429.200(b)), we propose in § 429.505(c) that a Primary Manufacturer has an ongoing obligation to report any updates to the information provided in proposed § 429.505(b) if the Primary Manufacturer becomes aware that any of such information has changed or is otherwise inaccurate. CMS will provide the Primary Manufacturer with a method to report any updates to the information provided in proposed § 429.505(b). For example, under proposed § 429.505(c), Primary Manufacturers must submit updates to the Primary Manufacturer's data submitted under proposed § 429.505(b) if the data was restated due to requirements of the Federal government entity that initially receives and oversees processing of such data. For example, under the Medicaid program, manufacturers must report revisions to best price under 42 CFR 447.510. The Primary Manufacturer must timely notify CMS if any such updates are applicable to the selected drug. This ongoing obligation to update the Primary Manufacturer's original data submissions is separate both from any voluntary submission of data from a Primary Manufacturer of a selected drug to inform renegotiation eligibility and selection (described at proposed § 429.615(a) and section II.G.4. of this proposed rule) and the required submission of data related to section 1194(e)(1) factors to CMS for drugs selected for renegotiation (described at proposed § 429.615(b)(1) and section II.G.4. of this proposed rule), which cover different reporting periods. As discussed further in section II.G.4.a. of this proposed rule, we may consider the Primary Manufacturer's prior data submission(s) under proposed § 429.505(b), including any updates to such information, to inform renegotiation eligibility and selection, and may use such information during renegotiation if a drug is selected for renegotiation.
b. Information Related to Section 1194(e)(2) Factors
Section 1194(e)(2) of the Act directs CMS, for purposes of negotiating the MFP for a selected drug with the Primary Manufacturer, to consider certain factors, as applicable to the selected drug, as the basis for determining its offers, as described at proposed § 429.510 and section II.F.3. of this proposed rule. These factors, listed at sections 1194(e)(2)(A) through (D) of the Act, include: (1) the extent to which the selected drug represents a therapeutic advance compared to existing therapeutic alternatives for the selected drug and the costs of such existing therapeutic alternatives; (2) FDA-approved prescribing information for the selected drug and its therapeutic alternatives; (3) comparative effectiveness of the selected drug and its
( printed page 36288)
therapeutic alternatives, including the effects of the selected drug and its therapeutic alternatives on specific populations (including individuals with disabilities, the elderly, the terminally ill, children, and other patient populations as described in section 1194(e)(2)(C) of the Act); and (4) the extent to which the selected drug and the therapeutic alternatives to the selected drug address unmet medical needs for a condition for which treatment or diagnosis is not addressed adequately by available therapy. Section 1194(e)(2) of the Act does not require the information listed to come from any specific source. As such, we considered various options for obtaining information related to the factors listed in section 1194(e)(2) of the Act, including an internal CMS analysis, obtaining the information from the Primary Manufacturer, obtaining information from the public, or some combination of these options. We believe that obtaining input from multiple perspectives, including but not limited to manufacturers, Medicare beneficiaries, academic experts, clinicians, caregivers, and other interested parties will provide the most comprehensive understanding of the selected drug and its therapeutic alternatives.
For section 1194(e)(2) factors, we propose to take a two-pronged approach consistent with policies for implementation for initial price applicability year 2028 as described in section 50.2 of Negotiation Program Guidance. In this approach, we would evaluate existing literature and real-world evidence, conduct internal analytics, and consult subject matter experts and clinicians, whether within CMS or external to CMS, when considering available evidence about alternative treatments to the selected drug (per the process proposed at § 429.510(b) and (c) and described in section II.F.3.c. of this proposed rule). We also intend to provide for a voluntary submission of information related to section 1194(e)(2) factors from manufacturers and the public through the Drug Price Negotiation ICR. Accordingly, consistent with policies for implementation for initial price applicability year 2028 as described in section 50.2 of the Negotiation Program Guidance, we propose at § 429.505(d) to codify that the submission of information related to section 1194(e)(2) of the Act will be open to the public and voluntary. We propose at § 429.505(d)(1) that any interested party, including the Primary Manufacturer of a selected drug, may submit information on selected drugs and their therapeutic alternatives (consistent with policies for implementation for initial price applicability year 2028 as described in section 60.3 of Negotiation Program Guidance) including information on whether the selected drug represents a therapeutic advance over its therapeutic alternative(s), prescribing information for the selected drug and its therapeutic alternative(s), comparative effectiveness data for the selected drug and its therapeutic alternative(s), information about the impact of the selected drug and its therapeutic alternative(s) on specific populations, information about patient experience, information on whether the selected drug addresses unmet medical need, or any combination thereof, as described in section 1194(e)(2) of the Act.
In accordance with section 1196(a)(4) of the Act, we also propose at § 429.505(d)(2) that the deadline for submission of information related to the factors listed in section 1194(e)(2) of the Act be the same date and time as the deadline for the Primary Manufacturer to submit non-FAMP and information related to factors listed in section 1194(e)(1) of the Act, that is by 11:59 p.m. PST on March 1 of the year of the selected drug publication date for the selected drug (described in proposed § 429.100(c) and section II.B.1. of this proposed rule). This serves to enable CMS to consider all submitted evidence in totality and meet the statutory deadline for the initial offer.
Section 1194(e)(2) of the Act also requires that CMS not use evidence from comparative clinical effectiveness research in a manner that treats extending the life of an individual who is elderly, disabled, or terminally ill as of lower value than extending the life of an individual who is younger, nondisabled, or not terminally ill. Accordingly, consistent with policies for implementation for initial price applicability year 2028 as described in section 50.2 of the Negotiation Program Guidance, we propose to codify at § 429.505(e), that we will not use evidence from comparative clinical effectiveness research in a manner that treats extending the life of an individual who is elderly, disabled, or terminally ill as of lower value than extending the life of an individual who is younger, nondisabled, or not terminally ill.
Specifically, we propose to codify at § 429.505(e)(1) that we will review cost-effectiveness measures used in studies relevant to a selected drug to determine whether the use of the measure is permitted in accordance with section 1194(e)(2) of the Act as described at § 429.505(e), as well as section 1182(e) of the Act and other applicable law, including section 504 of the Rehabilitation Act. We also propose at § 429.505(e)(2) that we may use content in a study that uses a cost-effectiveness measure if we determine that the cost-effectiveness measure used is permitted in accordance with section 1194(e)(2) of the Act as described at proposed § 429.505(e), as well as section 1182(e) of the Act and other applicable law, including section 504 of the Rehabilitation Act. In proposed § 429.505(e)(2), we propose that in instances where some, but not all, content in a study is excluded, we may still consider content that is relevant and allowable (for example, clinical effectiveness, risks, harms) under section 1194(e)(2) of the Act and section 1182(e) of the Act.
3. Methodology for Developing the Initial Offer (§ 429.510)
Section 1194(e) of the Act directs CMS to consider certain factors related to manufacturer-specific data and available evidence about therapeutic alternative(s) as the basis for determining offers and counteroffers in the negotiation process. Further, section 1194(b)(2)(B) of the Act requires CMS to provide the manufacturer of a selected drug with a written initial offer and a concise justification based on the factors described in section 1194(e) of the Act that were used in developing the offer. However, the statute does not specify how and to what degree each factor should be considered, thereby providing CMS with the discretion to determine how each factor is considered.
Consistent with section 1194(e) of the Act and with policies for implementation as described in section 60.3 of Negotiation Program Guidance, we propose in § 429.510 that for purposes of determining the initial offer, we would: (1) identify conditions for which the selected drug is used, as described in proposed § 429.510(a); (2) identify the therapeutic alternative(s), if any, for the selected drug, as described in proposed § 429.510(b) and (c) and consistent with policies for implementation as described in section 60.3.1 of Negotiation Program Guidance; (3) determine the starting point for the initial offer based on the price(s) of the therapeutic alternative(s) for the selected drug, if any, or an alternative price if there is no therapeutic alternative, as described in § 429.510(d) and consistent with policies for implementation as described in section 60.3.2 of Negotiation Program Guidance; (4) consistent with policies for implementation as described in section 60.3.3 of Negotiation Program Guidance
( printed page 36289)
and as proposed at § 429.510(e), evaluate the selected drug, including compared to its therapeutic alternative(s), for the purposes of adjusting the starting point using the negotiation factors outlined in section 1194(e)(2) of the Act, which would result in the preliminary price as defined at proposed § 429.20; and (5) adjust the preliminary price based on the negotiation factors outlined in section 1194(e)(1) of the Act and consistent with policies for implementation as described in section 60.3.4 of Negotiation Program Guidance as described in proposed § 429.510(f) to determine the initial offer price. In accordance with section 1194(b)(1) of the Act, this process will be conducted with the aim of achieving the lowest MFP for each selected drug.
In accordance with section 1194(b)(2)(F) of the Act we would not make any offers or accept any counteroffers for the MFP that are above the statutorily defined ceiling as indicated in proposed § 429.410 nor below the temporary floor for small biotech drugs, if applicable, as indicated in § 429.440.
a. Identifying Conditions for Which the Selected Drug Is Used (§ 429.510(a))
To identify conditions for which the selected drug is used, we propose at § 429.510(a)(1) to identify the FDA-approved indication(s) of the selected drug covered under Part D, payable under Part B, or both using prescribing information approved by the FDA for the selected drug, in accordance with section 1194(e)(2)(B) of the Act and consistent with policies for implementation as described in section 60.3.1 of Negotiation Program Guidance. In § 429.510(a)(2), we propose to consider off-label use to identify conditions for which the selected drug is used, if such use is included in evidence-based clinical practice guidelines and the off-label use is a medically accepted indication, as defined in section 1927(k)(6) of the Act, payable under Part B, covered under Part D, or both, taking into consideration the major drug compendia, authoritative medical literature, accepted standards of medical practice, or some combination thereof.
In proposed § 429.510(a)(3), we propose to exclude from our analysis for development of the initial offer any FDA-approved indication(s) or off-label uses for which we believe that utilization of the selected drug within such indication(s) and for such uses is intended solely for use in a setting in which the selected drug is not payable under Part B and not covered under Part D. Since the MFP resulting from a negotiation may only be applied for drugs payable under Part B, covered under Part D, or both, it is consistent for CMS to exclude from its analysis these FDA-approved indications or off-label uses. The MFP would not be applied unless payable under Part B or covered under Part D, so excluding such FDA-approved indications or off-label uses from CMS' analysis provides the opportunity to focus on indications for which the MFP would be applied when developing the initial offer. For example, we may not include an FDA-approved indication or off-label use in our analysis for the initial offer if such indication or off-label use is recommended for use in clinical practice guidelines only in inpatient hospital settings. We note that a Primary Manufacturer may suggest in its response to the initial offer that use within an FDA-approved indication or an off-label use that was not included in the initial offer is relevant for negotiating an MFP for the selected drug, in which case we may include such indication or off-label use in our consideration of any counteroffers or revised offers.
b. Identifying Therapeutic Alternatives for Each Condition (§ 429.510(b) and (c))
For each condition for which the selected drug is used, we would use the information identified in proposed § 429.510(b) and would follow the steps defined in proposed § 429.510(c) and consistent with policies for implementation as described in section 60.3.1 of Negotiation Program Guidance to identify a pharmaceutical therapeutic alternative(s), if any, for purposes of developing the initial offer. To identify potential therapeutic alternatives for the condition(s) for which a selected drug is used, we propose in § 429.510(b)(1) to use data submitted by the Primary Manufacturer and the public, prescribing information approved by the FDA, drug classification systems commonly used in the public and private sector for formulary development, major drug compendia, widely accepted clinical practice guidelines, evidence identified through the CMS-led literature review, published drug or drug class reviews, peer-reviewed studies, and Medicare claims or other data sets. In addition to brand name drugs and biological products, we propose to consider generic drugs and biosimilars, including specific formulations or dosage forms and strengths of a brand name drug, brand name biological product, generic drug, or biosimilar, as applicable, when identifying a potential therapeutic alternative(s) to a selected drug as described in proposed § 429.510(b)(2).
We considered evaluating non-pharmaceutical interventions as potential therapeutic alternatives to the selected drug, including requesting public comment on the use of health care services during the 45-day public comment period on the draft guidance for initial price applicability year 2028. Considering non-pharmaceutical interventions as a therapeutic alternative poses several challenges. First, from a clinical perspective, non-pharmaceutical interventions are not exclusively used as alternatives to drugs, or vice versa. For example, non-pharmaceutical interventions may serve as a complementary component of treatment provided at the same time as treatment with a drug. In other situations, such as cancer care, the standard of care may recommend the sequential use of non-pharmaceutical interventions and drugs, with the non-pharmaceutical intervention (for example, surgery) provided as a first-line treatment and then a drug that is used as the second-line treatment (for example, oral chemotherapy) or vice versa. Should a non-pharmaceutical intervention be considered a therapeutic alternative, such intervention must be an alternative to the drug rather than a complementary or sequential component of care, which may be difficult to clearly determine given the complex nature of treatment for many disease states and conditions. It may also be difficult to assess the comparative effectiveness of a non-pharmaceutical intervention and a drug. As a result of these considerations, we are not proposing to use non-pharmaceutical interventions as a therapeutic alternative for selected drugs in this proposed rule. As we continue to gain experience with the Negotiation Program, we may continue to evaluate approaches to the identification of therapeutic alternatives.
Section 1194(e)(2)(A) of the Act directs CMS to consider “[t]he extent to which such drug represents a therapeutic advance as compared to existing therapeutic alternatives and the costs of such existing therapeutic alternatives.” As such, we also considered how to understand this directive from statute if non-pharmaceutical interventions were included as therapeutic alternatives, including how to consider differential pricing between drugs and non-pharmaceutical interventions. Pricing and payment for drugs and services often differ substantially, not only within Medicare but also across other
( printed page 36290)
payer types, and non-pharmaceutical interventions may not have similar periodicity or frequency of administration as drugs, which could make comparisons challenging. If CMS were to consider non-pharmaceutical interventions as therapeutic alternatives, we would be required to adopt or create a methodology for determining the 30-day equivalent supply of a non-pharmaceutical intervention, which could include or exclude many components of a service. For example, if physical therapy could potentially serve as a therapeutic alternative to the use of a selected drug, we would need to determine how many and what types of physical therapy services and what duration of those services should be compared to the selected drug and over what time frame. Also, if a type of surgery could potentially serve as a therapeutic alternative to the use of the selected drug, we would need to determine what bundle of services from that surgery would be included in the therapeutic alternative. We believe that general rules would likely be insufficient to capture the heterogeneity of possible non-pharmaceutical interventions available and that specific analyses would be needed in each case. Therefore, we do not believe this is feasible at this time given the statutory timelines provided for negotiation and renegotiation.
Given these challenges, we believe that pharmaceutical therapeutic alternatives would be the most analogous alternative to the selected drug. By comparing a selected drug to a pharmaceutical therapeutic alternative, we would be able to consider the treatment effect and costs as directed by the statute in section 1194(e) of the Act while also operating within statutorily determined timeframes for the negotiation and renegotiation periods. We propose at § 429.510(c)(1) that we may consult with FDA to obtain information regarding other therapies with FDA-approved indications for the same condition. We may also consult with clinicians, patients or patient organizations, researchers, or any combination thereof to ensure that appropriate therapeutic alternatives are identified. We propose in § 429.510(c)(2)(i) to consider off-label use when identifying conditions for therapeutic alternatives if such use is included in evidence-based clinical practice guidelines and the off-label use is medically accepted and covered under Part D, payable under Part B, or both, taking into consideration the major drug compendia, authoritative medical literature, accepted standards of medical practice, or some combination thereof, as defined in proposed § 429.20.
We propose at § 429.510(c)(2)(ii) to identify potential therapeutic alternatives within the same pharmacologic class as the selected drug based on properties such as chemical class, therapeutic class, or mechanism of action and also propose at § 429.510(c)(2)(iii) to consider potential therapeutic alternatives in different pharmacologic classes based on our evaluation of the sources noted previously and in proposed § 429.510(b)(1). Where appropriate, only certain formulations or dosage forms and strengths of a brand name drug, brand name biological product, generic drug, or biosimilar (as described in § 429.510(b)(2)) would be identified as the therapeutic alternative to the selected drug as specified in proposed § 429.510(c)(2)(iv), for example, when one formulation of a drug meets the criteria for a therapeutic alternative, but other formulations do not.
In proposed § 429.510(c)(3), we propose that, in cases where there are many potential therapeutic alternatives for a given condition for which the selected drug is used, we may focus on a subset of therapeutic alternatives that are clinically comparable to the selected drug for the purpose of developing the initial offer. We note that when referencing a “therapeutic alternative” this may refer to one or more therapeutic alternative(s) or a subset of therapeutic alternatives that are clinically comparable. For example, for a potential therapeutic alternative, we may consider the place in therapy based on evidence-based clinical practice guidelines, pharmacologic and therapeutic characteristics, utilization in the Medicare population, and the availability of direct and indirect comparative evidence relative to the selected drug. In § 429.510(c)(4), we propose to prioritize clinical appropriateness in the selection of therapeutic alternatives.
c. Developing a Starting Point for the Initial Offer (§ 429.510(d))
To fulfill the statutory requirement to develop the initial offer per section 1194b(2)(B) of the Act in accordance with section 1194(e) of the Act, we propose at § 429.510(d) to determine a numerical starting point that would be adjusted based on the section 1194(e)(2) factors to determine a preliminary price (as described in § 429.510(e) and section II.F.3.d.1. of this proposed rule) which would then be adjusted based on the section 1194(e)(1) factors (as described in § 429.510(f) and section II.F.3.d.2. of this proposed rule). We considered several options for what price should be used as the starting point for developing the initial offer. Options considered included the use of the Part D net price(s), the ASP/WAC(s) of therapeutic alternative(s) for drugs covered under Part D or payable under Part B, respectively, or both; the unit cost of production and distribution for the selected drug; the ceiling for the selected drug (as described in proposed § 429.440(a)); a domestic reference price for the selected drug (for example, the Federal Supply Schedule [56]
(FSS) price); a “fair profit” price for the selected drug based on whether R&D costs have been recouped and margin on unit cost of production and distribution; Net Part D Plan Payment and Beneficiary Liability for drugs covered under Part D; the MFP, if available; a starting point between: (a) the Part B ASP/WACs, the Net Part D Plan Payment and Beneficiary Liability, or the combined Part B and D amount discussed previously for the therapeutic alternatives; and (b) the statutory ceiling; or considering a starting point between (a) the Part B ASPs/WACs, the Net Part D Plan Payment and Beneficiary Liability, or the combined Part B and Part D amount discussed previously for therapeutic alternatives and (b) unit cost of production and distribution of the selected drug; or some combination thereof. Under any of these options, the initial offer and final MFP would be capped at the statutory ceiling as required by section 1194(c) of the Act and proposed at § 429.440(b).
After considering these options and based on experience from implementation of the Negotiation Program for initial price applicability years 2026 through 2028, in accordance with section 1194(e)(2)(A) of the Act, which directs CMS to consider the cost of therapeutic alternative(s), we propose at § 429.510(d)(1) to use the price of the therapeutic alternative(s) to determine the starting point for developing the initial offer. We propose at § 429.510(d)(1)(i) that the price of a therapeutic alternative covered under Part D would be the lower of: the Net Part D Plan Payment and Beneficiary Liability, WAC, or the MFP for a selected drug negotiated for a prior
( printed page 36291)
initial price applicability year (regardless of whether the agreed-upon MFP for such selected drug has become effective). We propose at § 429.510(d)(1)(ii) that the price of a therapeutic alternative payable under Part B would be the lower of: ASP, the MFP for a selected drug negotiated for a prior initial price applicability year (regardless of whether the agreed-upon MFP for such selected drug has become effective), or WAC. We propose at § 429.510(d)(1)(iii) that for a therapeutic alternative that is both payable under Part B and covered under Part D, the price would be equal to a combined amount based on the price of the therapeutic alternative under Part D and the price under Part B (as determined under proposed § 429.510(d)(1)(i) and (ii)). We propose that the combined amount would be determined using an approach similar to the methodology used to combine the sum of the plan-specific enrollment weighted amounts and the payment amount under section 1847A(b)(4) of the Act; this single combined amount would then be weighted by utilization of the drug across Part B and Part D.
For a selected drug with no therapeutic alternative or for a selected drug where the price(s) of the therapeutic alternative(s) determined under proposed § 429.510(d)(1)(i) and (iii) are above the ceiling, we propose at § 429.510(d)(1)(iv) to use an alternative price equal to the lower of: the pharmaceutical price for the selected drug as included in the FSS as managed by the Department of Veterans Affairs per 48 CFR part 38 as most recently submitted by the Primary Manufacturer; or the maximum price a manufacturer can charge for the selected drug under 38 U.S.C. 8126 as most recently submitted by the Primary Manufacturer.
When determining the Net Part D Plan Payment and Beneficiary Liability of a therapeutic alternative, as defined in § 429.20, we would exclude PDE records for which a compound code indicates the PDE record is for a compounded drug as described in section II.B.5.a. of this proposed rule. We also believe it is important to reduce the total gross covered prescription drug costs by both DIR and Manufacturer Discount Program payments to permit an appropriate accounting of the price paid by the plan and beneficiary net of price concessions received by Part D plan or pharmacy benefit managers on behalf of a Part D plan. When assessing a therapeutic alternative(s) payable under Part B to determine a starting point for the initial offer, we believe using the lesser of ASP or WAC aligns with the payment amount under section 1847A(b)(4) of the Act, including in circumstances where the WAC of a therapeutic alternative is lower than its ASP. We also believe that considering the agreed-upon MFP for a selected drug that is payable under Part B, covered under Part D, or both, is also appropriate since the agreed-upon MFP may be the lowest available pricing metric for certain drugs.
In proposing this approach, we acknowledge that the therapeutic alternative(s) may not be priced to reflect the clinical benefit of the selected drug; however, using existing prices for therapeutic alternatives, including Net Part D Plan Payment and Beneficiary Liability, MFP, or WAC for a drug covered under Part D, or ASP, MFP, or WAC for a drug payable under Part B, enables CMS to start developing the initial offer within the context of the cost and clinical benefit of one or more drugs that treat the same disease or condition. By using the price(s) of the selected drug's therapeutic alternative(s), we would be able to focus the adjustments on section 1194(e)(2) factors by adjusting this starting point (as described in proposed § 429.510(e)(4)) based on the overall evidence of benefits and harms offered by the selected drug as compared with its therapeutic alternative(s). The other options considered do not provide a starting point that reflects the cost of therapeutic alternatives in the current market, which is an important factor when considering the overall benefit that a treatment brings to Medicare beneficiaries relative to the other drug(s) available to treat the patient's disease or condition.
We propose at § 429.510(d)(2) that the prices determined under § 429.510(d)(1) and the starting point would be expressed as a 30-day equivalent supply. We propose at § 429.510(d)(2)(i) to use the same methodology described in proposed § 429.410(b)(1) to calculate the 30-day equivalent supply for drugs covered under Part D, as appropriate, unless we determine it is appropriate to apply an alternative methodology as described further below. We propose at § 429.510(d)(2)(ii) to use the same methodology described in proposed § 429.410(b)(2) to calculate the 30-day equivalent supply for drugs payable under Part B, as appropriate, unless we determine it is appropriate to apply an alternative methodology as described further below. We propose at § 429.510(d)(2)(iii) to calculate the 30-day equivalent supply of a therapeutic alternative that is both covered under Part D and payable under Part B separately (as determined in proposed § 429.510(d)(1)(i) and (ii)), unless we determine it is appropriate to apply an alternative methodology as discussed below. We also propose in § 429.510(d)(2)(iii) that we would determine the 30-day equivalent supply for the price of the therapeutic alternative that is payable under Part B and, separately determine the 30-day equivalent supply for the therapeutic alternative that is covered under Part D prior to combining these amounts using the methodology described in proposed § 429.415(d)(1)(iii) to result in a single combined price expressed as a 30-day equivalent supply.
In certain circumstances it may be necessary to use an alternative methodology to calculate a 30-day equivalent supply, for example, if a therapeutic alternative for a condition is typically prescribed for a period meaningfully shorter than 30 days (for example, for a two-week period, meaning that one fill would be defined as a 30-day equivalent supply despite lasting only two weeks), and the selected drug does not have a similar prescribing pattern, we may use an alternative methodology to calculate 30-day equivalent supply for the therapeutic alternative to ensure that its price is expressed on comparable terms to a 30-day equivalent supply. In circumstances such as these, we propose at § 429.510(d)(4)(iv) to use a tailored alternative methodology to calculate the 30-day equivalent supply for therapeutic alternative(s) covered under Part D, payable under Part B, or both, when appropriate. The tailored methodology would promote comparability between the therapeutic alternative(s) and the selected drug.
We propose in § 429.510(d)(3) to use the price(s) identified under proposed § 429.510(d)(1) to determine the starting point for developing the initial offer. We propose at § 429.510(d)(3)(i) that if there is no therapeutic alternative for a selected drug or there is no price of the therapeutic alternative(s) determined under proposed § 429.510(d)(1)(i) through (iii) that are below the ceiling, then the starting point for developing the initial offer is the lower of the prices listed at proposed § 429.510(d)(3)(i)(A) through (C). That is, we propose to use the lower of the maximum price a manufacturer can charge under 38 U.S.C. 8126 as most recently submitted by the Primary Manufacturer, the pharmaceutical price for the selected drug as included in the FSS as managed by the Department of Veterans Affairs per 48 CFR part 38 (hereinafter the “FSS price”) as most recently submitted by the Primary Manufacturer, or the ceiling.
( printed page 36292)
If there are multiple therapeutic alternatives and at least one therapeutic alternative price identified is below the ceiling, we propose at § 429.510(d)(3)(ii) to determine a starting point for developing the initial offer within a range based on the lower of the prices of the therapeutic alternatives determined under § 429.510(d)(1) and the ceiling. In implementing this proposal, we may weigh prices used to determine the range based on utilization, for example, by the utilization of therapeutic alternatives within and across multiple conditions or other patterns of use for the therapeutic alternatives or the selected drug. When determining the starting point within this range, we may consider the therapeutic alternative prices for each condition if such prices are available and if such prices are different across conditions.
Finally, we propose at § 429.510(d)(3)(iii) that if there is one therapeutic alternative for the selected drug with a price that is below the ceiling, the price of such therapeutic alternative is the starting point. In all cases, the starting point would not exceed the statutory ceiling and would be subject to adjustments as described further in proposed § 429.510(e) and section II.F.3.d. of this proposed rule.
d. Adjusting the Starting Point and Preliminary Price Based on the Factors Listed at Section 1194(e) of the Act (§ 429.510(e) and (f))
Section 1194(e) of the Act directs CMS to consider the factors listed at section 1194(e)(1) and 1194(e)(2) of the Act as the basis for any offers and counteroffers and, in accordance with section 1194(b)(1) of the Act, CMS must develop and use a consistent methodology and process for negotiations to achieve the lowest MFP for each selected drug. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, sections 60.3.3 and 60.3.4 of the Negotiation Program Guidance with respect to initial price applicability year 2028. With respect to initial price applicability year 2029 and subsequent years, and consistent with policies for implementation as described in sections 60.3.3 and 60.3.4 of the Negotiation Program Guidance, we are proposing at § 429.510(e) to adjust the starting point determined under proposed § 429.510(d), based on the section 1194(e)(2) factors to determine the preliminary price and then adjusting the preliminary price based on the section 1194(e)(1) factors, as described in § 429.510(f), to determine the initial offer. This approach ensures that we consider each section 1194(e) factor while also providing a consistent methodology for doing so.
(1) Adjusting the Starting Point Based on Section 1194(e)(2) Factors (§ 429.510(e))
To evaluate the section 1194(e)(2) factors, including the clinical benefit conferred by the selected drug compared to its therapeutic alternative(s), we propose at § 429.510(e)(1) to use a qualitative approach to broadly evaluate the body of available evidence, including information received from the public and the Primary Manufacturer as described in proposed §§ 429.505(b), 429.505(d), and 429.515(b), evidence identified through a CMS-led literature review, Medicare claims or other datasets, potentially including evidence related to health care resource utilization and usage patterns of the selected drug versus its therapeutic alternative(s), clinical data, or other information relevant to the selected drug and its therapeutic alternative(s). We also propose at § 429.510(e)(2) that we may consult with clinicians, patients or patient organizations, researchers, and/or FDA. This review would be complementary to additional engagement opportunities for interested parties—specifically, meetings with the Primary Manufacturer and public events as proposed in § 429.515 and described in section II.F.4.b. of this proposed rule—after the March 1 deadline described at proposed § 429.505(d)(2) for submission of section 1194(e)(2) data.
As a complement to the public submission of information on section 1194(e)(2) factors and consistent with policies for implementation as described in section 50.2 of Negotiation Program Guidance, we propose at § 429.510(e)(1) to additionally evaluate existing literature and real-world evidence, conduct internal analytics, and consult subject matter experts and clinicians on these topics when considering available evidence about alternative treatments to the selected drug. When evaluating the literature from the public and manufacturer submissions as well as literature from CMS' review, we intend to consider the source, rigor of the study methodology, current relevance to the selected drug and its therapeutic alternative(s), whether the study has been through peer review, study limitations, degree of certainty of conclusions, risk of bias, study time horizons, generalizability, study population, and relevance to the negotiation factors listed in section 1194(e)(2) of the Act to ensure the integrity of the contributing data within the negotiation process. We also propose to prioritize research, including both observational research and research based on randomized samples, that is methodologically rigorous, appropriately powered (that is, has sufficient sample size) to answer the primary question of the research, and structured to avoid potential false positive findings due to multiple subgroup analyses. We propose to consider research and real-world evidence relating to Medicare populations, including individuals with disabilities, patients with end-stage renal disease, and Medicare-aged populations, as particularly important. In considering impact on specific populations and patients with unmet medical needs, we intend to prioritize research specifically designed to focus on these populations over studies that include outcomes for these populations but for which these populations were not the primary focus.
This approach would provide a pathway for CMS to consider the multitude of information expected from public input, including but not limited to peer-reviewed research, expert reports or white papers, clinician expertise, real-world evidence, and patient experience. This approach also would provide us with the flexibility to consider a variety of aspects in our evaluation of comparative effectiveness, including patient experiences, disease severity, treatment complexity, and/or other unique considerations related to use of the selected drug or therapeutic alternatives.
To consider comparative effectiveness of a selected drug and its therapeutic alternative(s) as required by section 1194(e)(2)(C) of the Act, we propose at § 429.510(e)(3) to identify outcomes to evaluate for each identified condition for which the selected drug is used. Outcomes of interest may include direct clinical outcomes (for example, cure, mortality) or validated or reasonably likely surrogate endpoints (for example, serum hemoglobin A1c) or both. In determining outcomes of interest, we would consider patient-reported outcomes and outcomes of importance to patients, if available. We would consider the identified outcomes of interest, including patient-centered outcomes, and patient experience data, when evaluating the clinical benefit of the selected drug and its therapeutic alternative(s) for those conditions. In addition, we propose at § 429.510(e)(3)(i)(C) that outcomes and contextual factors such as health-related
( printed page 36293)
quality of life or patient and caregiver preferences would also be considered to the extent these outcomes and factors correspond with benefits or harms to the individuals taking the selected drug or therapeutic alternative(s) and are appropriately measurable and quantifiable.
We propose at § 429.505(e) that when evaluating such information we would not, per section 1194(e)(2) of the Act, use evidence in a manner that treats extending the life of an individual who is elderly, disabled, or terminally ill as lower value than extending the life of an individual who is younger, non-disabled, or not terminally ill, and would not, per section 1182(e) of the Act, use QALYs. Outcomes of interest may include direct clinical outcomes (for example, cure, mortality) and/or validated or reasonably likely surrogate endpoints (for example, serum hemoglobin A1c). In determining outcomes of interest, we would consider patient-reported outcomes and outcomes of importance to patients, if available. We may also consider additional outcomes and contextual factors, such as health-related quality of life or patient/caregiver preferences regarding treatment, to the extent these outcomes and factors correspond with benefits or harms to individuals taking the selected drug or therapeutic alternatives. In proposed § 429.510(e)(3)(i)(D), we propose to also consider the caregiver perspective to the extent that it reflects directly upon the experience or relevant outcomes of the patient taking the selected drug. As described in proposed § 429.510(e)(3)(ii), relevant outcomes would be identified using the CMS-led literature review and information submitted by manufacturers and the public, including patients and caregivers, as well as in the public events described in proposed § 429.515(b).
In all cases, we propose to consider applicable evidence and other input collectively, within the context of the course of care for each condition for which the selected drug is used. As noted previously, we believe this approach would provide flexibility to consider a variety of aspects in our evaluation of comparative effectiveness, including patient experiences, disease severity, treatment complexity, and/or other unique considerations related to the use of the selected drug or its therapeutic alternative(s) for a given condition.
Once the starting point for the initial offer has been established and evidence on section 1194(e)(2) factors has been considered, we would consider the information regarding the four factors outlined in section 1194(e)(2) of the Act collectively and within the context of the course of care for each condition for which the selected drug is used as described at proposed § 429.510(e)(4) and apply an upward adjustment, downward adjustment, or no adjustment to the starting point to determine the preliminary price as described at proposed § 429.510(e)(5). In accordance with section 1194(b)(1) of the Act, consideration and adjustment for the section 1194(e) factors will be made with the aim of achieving the lowest MFP for each selected drug. We considered employing both a qualitative approach (for example, adjusting the starting point upward or downward relative to the section 1194(e)(2) factors offered by the selected drug compared to its therapeutic alternative(s)) and a more thoroughly pre-specified quantitative approach. Consistent with the first three cycles of negotiation, we are proposing to use a qualitative approach to consider nuanced differences between drugs, for example, interactions with other treatments commonly prescribed simultaneously for a condition or disease, and other factors that might not be captured in a more thoroughly pre-specified quantitative approach.
(a) Analysis for Selected Drugs With Therapeutic Alternative(s)
For each condition for which we identify a therapeutic alternative, we propose to consider the information regarding the four factors outlined in section 1194(e)(2) of the Act collectively. As described at proposed § 429.510(e)(4)(i), we propose that such review would include, but is not limited to, examining improvements (as described at proposed § 429.510(e)(4)(i)) in outcomes (as determined in proposed § 429.510(e)(3)) to determine the extent to which a selected drug represents a therapeutic advance (as defined at proposed § 429.20) as compared to its therapeutic alternative(s) (as defined at proposed § 429.20) (for example, if selected drug is curative versus a therapeutic alternative that delays progression) and would consider the costs of the selected drug and its therapeutic alternative(s) as described at proposed § 429.510(e)(4)(i)(A).
We propose at § 429.510(e)(4)(i)(B) to consider the magnitude of differences in outcomes of interest conferred by the selected drug compared to the selected drug's therapeutic alternative(s) for each condition in which we identify a therapeutic alternative, when determining the extent to which a selected drug represents a therapeutic advance. We understand that a selected drug can be first in class.[57]
However, other drugs may have become available since the selected drug's initial approval and therefore we propose in the definition of “therapeutic advance” at proposed § 429.20 to consider the extent to which a selected drug represents a therapeutic advance for a condition at the time the section 1194(e)(2) data is submitted, which is the date specified in proposed § 429.505(d)(2). In accordance with section 1194(e)(2)(A) of the Act, we propose to review the analyses detailed previously for each identified condition of the selected drug and its therapeutic alternative(s) to determine the extent to which the selected drug represents a therapeutic advance as compared to its therapeutic alternative(s) as described in proposed § 429.510(e)(4)(i). For purposes of the Negotiation Program, anytime CMS considers therapeutic advance, CMS proposes to consider the extent to which the drug represents a therapeutic advance at the time of consideration based on all available information at such time of consideration.
In accordance with section 1194(e)(2)(C) of the Act, we would also consider the effects of the selected drug and its therapeutic alternative(s) on specific populations, such as individuals with disabilities, the elderly, individuals who are terminally ill, children, and other patient populations. To do so, we propose to identify studies, if available, focused on the conditions for which each selected drug is used and the impact of the selected drug and its therapeutic alternative(s) on such specific populations. Further, per section 1194(e)(2)(D) of the Act, for each condition in which CMS has identified a therapeutic alternative, we would consider the extent to which the selected drug and its therapeutic alternative(s) address an unmet medical need for each condition for which the selected drug is used. For purposes of the Negotiation Program, anytime CMS considers an unmet medical need, CMS would consider the extent to which the drug addresses an unmet medical need at the time of consideration based on all available information at such time of consideration. When considering unmet medical need (see proposed § 429.20), we propose to consider the selected drug, its therapeutic alternative(s), if
( printed page 36294)
any, and any existing treatment options, which may include pharmacologic or non-pharmacologic treatments. We may consider the nonbinding recommendations in the FDA's “Guidance for Industry Expedited Programs for Serious Conditions—Drugs and Biologics,” [58]
as well as any updates that may be issued by FDA in the future, when determining the extent to which a selected drug addresses an unmet medical need for a condition.
(b) Analysis for Conditions of Selected Drugs Without Therapeutic Alternatives
For conditions of selected drugs without therapeutic alternatives, we would consider the information regarding the four factors outlined in section 1194(e)(2) of the Act collectively. We propose at § 429.510(e)(4)(ii) certain parameters for our review of conditions of selected drugs where we need to account for the lack of therapeutic alternatives for such conditions. Specifically, we propose at § 429.510(e)(4)(ii)(A) that for each condition for which a selected drug is used but does not have a therapeutic alternative, we would consider the totality of available information relevant to the section 1194(e)(2) factors as detailed previously and in proposed § 429.510(e)(1), including information received from Primary Manufacturers and the public as described in proposed § 429.505(d), and evidence identified through a CMS-led literature review. We also propose at § 429.510(e)(4)(ii)(B) to consider the selected drug and any existing treatment options, which may include pharmacologic or non-pharmacologic treatments, to determine the extent to which the selected drug addresses an unmet medical need (as defined in § 429.20) at the condition level. For purposes of the Negotiation Program, anytime CMS considers an unmet medical need, CMS would consider the extent to which the drug addresses an unmet medical need at the time of consideration based on all available information at such time of consideration. We would consider unmet medical need in the same manner as discussed in section II.F.3.a. of this proposed rule. At proposed § 429.20, we propose to define unmet medical need as a circumstance in which the relevant condition is one for which no treatment options exist, or existing treatments do not adequately address the condition. As noted previously, we may consider the nonbinding recommendations in the FDA “Guidance for Industry Expedited Programs for Serious Conditions—Drugs and Biologics,” as well as any updates that may be issued by FDA in the future, when considering the extent to which a drug addresses an unmet medical need for the purpose of the Negotiation Program.
Finally, as described at proposed § 429.510(e)(4)(ii)(C), we propose to examine improvements in outcomes, such as the magnitude of differences in outcomes of interest conferred by the selected drug, for a condition wherein CMS has not identified a therapeutic alternative to determine the extent to which a selected drug represents a therapeutic advance. For purposes of the Negotiation Program, anytime CMS considers therapeutic advance, CMS would consider the extent to which the drug represents a therapeutic advance at the time of consideration based on all available information at such time of consideration
(c) Preliminary Price
As noted in section II.F.3.d.(1).(a). of this proposed rule, we propose to take a qualitative approach to adjusting the starting point based on the unique characteristics of the drug and its therapeutic alternative(s), if any, as well as the patient population(s) taking the selected drug. For each selected drug, we propose to adjust the applicable starting point (as determined in proposed § 429.510(d)) upward or downward or to not adjust (as described in proposed § 429.510(e)(5)) based on the totality of the relevant information and evidence submitted and gathered through our analysis based on section 1194(e)(2) factors. We may adjust the starting point based on how the section 1194(e)(2) factors apply with respect to individual condition(s) in cases where there are notable differences relative to the therapeutic alternative(s).
After the starting point is adjusted, if applicable, as described in proposed § 429.510(e)(4), as appropriate, based on section 1194(e)(2) factors, evaluated using data submitted by the Primary Manufacturer and the public through the Drug Price Negotiation ICR and gathered through our analyses and literature review, the resulting price is referred to as “the preliminary price” (defined in proposed § 429.20).
We propose to adjust the preliminary price, as appropriate, based on data submitted by the Primary Manufacturer in accordance with section 1194(e)(1) of the Act, as described in detail in section II.F.3.d.(2). of this proposed rule and proposed § 429.510(f).
(2) Adjusting the Preliminary Price Based on Consideration of Section 1194(e)(1) Factors
Section 1194(e)(1) of the Act directs CMS to consider certain factors, which must be reported by each Primary Manufacturer, when determining offers and counteroffers. To fulfill this requirement, we propose at § 429.510(f) to adjust the preliminary price based on the factors listed at section 1194(e)(1) of the Act. The preliminary price may be adjusted upward, adjusted downward, or not adjusted to account for these manufacturer-specific data elements. In accordance with section 1194(b)(1) of the Act, this process would be conducted with the aim of achieving the lowest MFP for each selected drug. The section 1194(e)(1) factors are listed at proposed § 429.505(b)(2) and include: (1) R&D costs of the manufacturer for the drug and the extent to which the manufacturer has recouped R&D costs; (2) current unit costs of production and distribution of the drug; (3) prior Federal financial support for novel therapeutic discovery and development with respect to the drug; (4) data on pending and approved patent applications or exclusivities recognized by the FDA, and applications and approvals under section 505(c) of the FD&C Act or section 351(a) of the PHS Act for the drug; and (5) market data and revenue and sales volume data for the drug in the United States.
We propose to consider the five factors outlined in section 1194(e)(1) of the Act in totality at proposed § 429.510(f)(1) and apply an upward adjustment, downward adjustment, or no adjustment to the preliminary price as described at proposed § 429.510(f)(2). We provide illustrative examples of how we might adjust, or not adjust, the preliminary price based on evaluation of a manufacturer-specific data element later in this section. However, the overall adjustment, inclusive of all five elements taken together, may differ from the example adjustment for any single element viewed in isolation.
Section 1194(e)(1)(A) of the Act requires CMS to consider the extent to which the Primary Manufacturer has recouped its R&D costs. As an example of how we could approach this consideration, CMS could compare the R&D costs with the global and U.S. net revenue for the selected drug reported by the Primary Manufacturer to determine the extent to which the Primary Manufacturer has recouped its R&D costs. For example, if a Primary Manufacturer has not recouped its R&D costs, we may consider adjusting the preliminary price upward. Conversely,
( printed page 36295)
if a Primary Manufacturer has recouped its R&D costs, we may consider adjusting the preliminary price downward or apply no adjustment. We may use the R&D costs reported by the Primary Manufacturer and the calculated recouped costs, including the assumptions and calculations in the accompanying narrative text, and/or other factors as described in proposed § 429.505(b)(2) to adjust the preliminary price.
As an example for how we may consider the relationship between the preliminary price and the unit costs of production and distribution (the factor listed at section 1194(e)(1)(B) of the Act), we may consider adjusting the preliminary price downward if the unit costs of production and distribution are lower than the preliminary price, or upward if the unit costs of production and distribution are greater than the preliminary price. Again, we may consider the assumptions and calculations in the accompanying narrative text submitted by the Primary Manufacturer of the selected drug as described in proposed § 429.505(b) to determine if an adjustment is appropriate.
As an example of how we may consider the extent to which the Primary Manufacturer benefited from Federal financial support with respect to the selected drug (the factor listed at section 1194(e)(1)(C) of the Act), we may consider adjusting the preliminary price downward if funding for the discovery and development of the drug was received from Federal sources.
We would also review the patents and exclusivities reported as we develop our initial offer, consistent with section 1194(e)(1)(D) of the Act. We believe that this information would support CMS' consideration of the factors listed at section 1194(e) of the Act and in proposed § 429.505(b)(2) and (d)(3). For instance, patents and exclusivities may inform our understanding of therapeutic alternatives and other available therapy for the purposes of adjusting for clinical benefit, including consideration of the extent to which the selected drug represents a therapeutic advance or the extent to which the selected drug addresses an unmet medical need. More specifically, in light of exclusivities, there may be no other available therapy aside from the selected drug that adequately addresses treatment or diagnosis of a disease or condition, and consideration of such information would be relevant to our consideration of the extent to which the selected drug addresses an unmet medical need for that disease or condition.
Finally, we would consider how the market data and revenue and sales volume data compare to the preliminary price in accordance with section 1194(e)(1)(E) of the Act. For example, if the average commercial net price is lower than the preliminary price, we may consider adjusting the preliminary price downward. If the average commercial net price is greater than the preliminary price, we may consider adjusting the preliminary price upward.
We propose in § 429.510(f)(3) that after any adjustments to the preliminary price are made as described in paragraphs (f)(1) and (f)(2), the result is the initial offer unless paragraph (f)(3)(i) applies. As proposed in § 429.510(f)(3)(i), if the resulting amount is above the ceiling (as determined in § 429.410), then the initial offer will be equal to the ceiling, and if the amount is below the temporary floor for small biotech drugs (as determined in proposed § 429.440), if applicable, then the initial offer will be equal to the temporary floor.
4. Engagement With Primary Manufacturers and Interested Parties (§ 429.515)
a. Engagement With Primary Manufacturers
Consistent with policies for implementation as described in section 60.4 of the Negotiation Program Guidance, we propose in § 429.515 to hold up to four, optional meetings in a form and manner specified by CMS with Primary Manufacturers of selected drugs that have submitted the information set forth in proposed § 429.505. As proposed in § 429.515(a)(1), the first meeting that we would offer Primary Manufacturers to attend would be intended for the Primary Manufacturer to provide additional context on their data submission of the section 1194(e)(1) factors and section 1194(e)(2) factors described in proposed § 429.505(b)(2) and (d)(3), respectively, as we begin evaluating the data submission and developing an initial offer as described in proposed § 429.510. We would also offer Primary Manufacturers the opportunity to attend up to three optional meetings that would focus on the section 1194(e)(1) factors and section 1194(e)(2) factors, and other topics aimed at working toward an agreement on an MFP. During these meetings, discussion of disputes and program policies regarding the negotiation process would be considered out of scope. As proposed in § 429.515(a)(2), meetings would be attended solely by representatives of the Primary Manufacturer and of CMS. The number of attendees would be limited as specified by CMS via future communications with the Primary Manufacturer specific to each initial price applicability year. For example, as described in section 60.4.4 of the Negotiation Program Guidance, CMS and the Primary Manufacturer were permitted to bring up to eight meeting attendees and both parties would share their participant lists ahead of each meeting. We determined this meeting attendee number after considering the roles from each party that would be critical to the conversation while ensuring that the meeting is sized appropriately to encourage active discussion. Given these considerations, we propose at § 429.515(a)(2) to continue to limit the number of meeting attendees.
We propose at § 429.515(a)(3)(i) that Primary Manufacturers may share new information on section 1194(e)(2) factors during meetings. The Primary Manufacturer may bring materials to facilitate discussion which must comply with limits on the amount and format of such materials as specified by CMS, and we may request that the Primary Manufacturer provide copies of any presented or discussed materials after the meeting in which they are presented or discussed, as proposed in § 429.515(a)(3)(ii). For example, for initial price applicability year 2028, if a Primary Manufacturer is interested in sharing materials at a negotiation meeting, such materials are limited to 15 pages (or a combination of pages, slides, and/or charts and graphs totaling 15 pages) and no more than 30 citations, to focus the discussion on issues that can reasonably be discussed within the scope of the meeting. We would specify the limits on the amount and format of materials specific to each initial price applicability year via future communications with the Primary Manufacturer. We propose at § 429.515(a)(1)(ii) that new data related to section 1194(e)(1) factors, as described in proposed § 429.505(b), would not be considered. Rather, any information shared during these meetings and materials shared afterwards should only contextualize the Primary Manufacturer's submission of data related to section 1194(e)(1) factors specified in proposed § 429.505(b).
We propose at § 429.515(a)(4) for the first optional meeting that CMS would offer to Primary Manufacturers to attend would occur, at a time to be specified by CMS, after the data submission deadline specified in section II.F.2. of this proposed rule and in proposed § 429.505(b)(1) and (d)(2) and before the
( printed page 36296)
provision of CMS' initial offer, as set forth in proposed § 429.520. We would offer up to three additional optional meetings to occur after the provision of CMS' initial offer. If accepted by the Primary Manufacturer, such meeting(s) would occur, at a time to be specified by CMS, after the provision of CMS' initial offer as set forth in proposed § 429.520 and before the provision of CMS' final offer, if applicable, as specified in proposed § 429.535. A written record of these meetings would be developed and retained by CMS in compliance with applicable Federal laws, including the Federal Managers' Financial Integrity Act and the Federal Records Act, and would be subject to the confidentiality policy described in section II.D.1. of this proposed rule and in proposed § 429.300. The Primary Manufacturer may also develop and retain its own written record. As proposed in § 429.515(a)(5), audio or video recording of the meetings would not be permitted.
As described in section II.D.1. of this proposed rule regarding proposed § 429.300, we would not publicly discuss ongoing negotiations with a Primary Manufacturer, including details of the negotiation meetings. A Primary Manufacturer may publicly disclose information regarding ongoing negotiations with CMS at their discretion. If a Primary Manufacturer discloses information regarding any aspects of the negotiation process prior to the explanation for the MFP being released by CMS, we reserve the right to publicly discuss the specifics of the negotiation process regarding that Primary Manufacturer.
b. Engagement With Interested Parties
At proposed § 429.515(b), we are proposing to codify the policy as described in section 60.4.1 of the Negotiation Program Guidance to hold events in a form and manner and at times to be specified by CMS with interested parties to seek input from patients and other interested parties about selected drugs and therapeutic alternatives. These public engagement events are intended to bring together patient-focused interested parties to share feedback with us on patient experiences with the conditions or diseases treated by the selected drug, as well as with the selected drugs and therapeutic alternatives to the selected drugs, and other information as we review data related to section 1194(e)(2) factors and develop an initial offer for each selected drug. These public engagement events allow us to hear from patients and other stakeholders close to the patient experience—directly and in their own words—about patients' personal experiences and perspectives on their condition(s) and about the drug(s) used to treat those conditions. This type of information helps inform CMS' understanding of what matters to patients. We would use the information shared during these patient-focused events to better understand patients' experiences with the conditions and diseases treated by the selected drug and their experiences with the selected drugs themselves, as well as to inform CMS' identification of therapeutic alternatives, key outcomes, and adjustment of the starting point to develop the initial offer. These events would be held annually in the Spring.
Public engagement events for each initial price applicability year may include, for example, patient-focused roundtable events that would be open to patients, patient advocacy organizations, and caregivers and would allow for discussion among speakers. These patient-focused roundtable events may focus on one selected drug or group selected drugs by condition when appropriate as determined by CMS.
The public engagement events also may include one town hall meeting for all selected drugs that would be focused on the clinical considerations related to the selected drugs and would be open to practicing clinicians and researchers, as well as other interested parties. This town hall meeting may be divided into multiple sessions and may be held across multiple days. We may have the opportunity to ask follow-up questions of participants at the town hall meeting.
Lastly, we may incorporate drugs selected for renegotiation into the public engagement events for drugs selected for negotiation or we may hold separate events specifically for drugs selected for renegotiation.
5. Provision of CMS' Initial Offer and Concise Justification (§ 429.520)
Section 1194(b)(2)(B) of the Act requires that, not later than June 1 following the selected drug publication date with respect to the initial price applicability year, CMS shall provide the Primary Manufacturer of the selected drug with a written initial offer that contains the proposal for the MFP of the drug and a concise justification based on the factors described in section 1194(e) that were used in developing such offer. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for example, section 60.4 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, and consistent with policies for implementation as described in section 60.4 of the Negotiation Program Guidance, we are proposing in § 429.520(a) that the written initial offer from CMS would be provided to the Primary Manufacturer no later than June 1 following the selected drug publication date as defined in proposed § 429.20. We propose at § 429.520(a)(1) that this written initial offer would be accompanied by an Addendum to the Negotiation Program Agreement populated with the proposal for the MFP for CMS and the Primary Manufacturer to formalize agreement upon the MFP if such agreement is reached at this stage. Consistent with proposed § 429.410(a), no written initial offer can exceed the statutorily determined ceiling as defined in section 1194(c) of the Act and proposed in section II.E.3. of this proposed rule and in proposed § 429.410(b). Consistent with proposed in § 429.440(b), no written initial offer can be less than the Temporary Floor for Small Biotech Drugs, if applicable, as defined in section 1194(d) of the Act and proposed in section II.E.9. of this proposed rule.
With respect to initial price applicability year 2029 and subsequent years, we are proposing in § 429.520(b) that CMS would include a concise justification for the written initial offer based on the data set forth in proposed § 429.505. Consistent with proposed § 429.505(a), CMS considers the Primary Manufacturer-required data specified in § 429.505(b)(2) and evidence about the selected drug and therapeutic alternatives specified in § 429.505(d)(3), as applicable to the drug, as the basis for determining the offers and counteroffers for the selected drug. As proposed in § 429.520(b)(1), the concise justification would include a qualitative description of the factors from section 1194(e) of the Act as proposed in § 429.505(b) and (d) and a description of the methodology that CMS used to develop the written initial offer as proposed in section II.F.3. of this proposed rule and under proposed subpart F. As proposed in § 429.520(b)(2), the information contained in the concise justification would provide the Primary Manufacturer with information on the range of evidence and other information considered under section 1194(e) of the Act that CMS found compelling during the development of the written initial offer, and may include information obtained through events with interested parties as proposed in § 429.520(b)(3). We believe the information in the
( printed page 36297)
concise justification would provide the Primary Manufacturer with information to build a statutory written counteroffer if the Primary Manufacturer decides to reject the written initial offer.
The written initial offer and concise justification would not include information that we determine to be third-party proprietary pricing information, information that could lead to the calculation of a third-party's proprietary pricing information, PHI/PII, other information that is protected from disclosure under other applicable law in accordance with the confidentiality policies described in section II.D.1. of this proposed rule and in proposed § 429.300, or the starting point.
6. Statutory Written Counteroffers (§ 429.525)
Section 1194(b)(2)(C) of the Act requires that the Primary Manufacturer shall either accept the written initial offer under section 1194(b)(2)(B) of the Act, or propose a counteroffer, within 30 days of receipt of the written initial offer. If a Primary Manufacturer proposes a counteroffer, such counteroffer shall be in writing and shall be justified based on the factors described in section 1194(e) of the Act. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 60.4 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, and consistent with policies for implementation as described in section 60.4 of the Negotiation Program Guidance, we are proposing in proposed § 429.525(a) that the Primary Manufacturer would have no more than 30 days from receipt of the written initial offer from CMS to respond in writing by either accepting the initial offer for the selected drug or making a statutory written counteroffer and providing a justification for such counteroffer based on the data described in proposed § 429.505(b) and (d). As proposed in § 429.525(b)(1)(ii), any statutory written counteroffer must respond to the concise justification provided in CMS' written initial offer, and the Primary Manufacturer's response must focus on the factors described in section 1194(e) of the Act, as set forth in proposed § 429.505(b) and (d). We propose at § 429.525(b)(1)(iii) that the statutory written counteroffer justification must indicate the reasons the Primary Manufacturer believes that the information submitted by the Primary Manufacturer on the section 1194(e)(1) factors or section (e)(2) factors, or other available data related to the selected drug and its therapeutic alternative(s), as described in section 1194(e)(2) of the Act, supports the Primary Manufacturer's statutory written counteroffer or otherwise does not support CMS' written initial offer. Primary Manufacturers may also include in their statutory written counteroffer justification new information regarding the selected drug and its therapeutic alternative(s) as described in section 1194(e)(2) of the Act that supports the counteroffer.
We propose at § 429.525(b)(1)(i) that the Primary Manufacturer must provide a proposal for the MFP for the selected drug in its statutory written counteroffer. The proposal for the MFP must be a single price for the selected drug per 30-day equivalent supply, weighted across dosage forms and strengths. For a statutory written counteroffer to be considered complete, we propose at § 429.525(b)(2) that a Primary Manufacturer must complete an Addendum to the Negotiation Program Agreement, as described in section II.C.1. of this proposed rule and in proposed § 429.200(e), in the CMS HPMS and must submit the statutory written counteroffer in a form and manner specified by CMS as part of the Drug Price Negotiation ICR. A completed Addendum to the Negotiation Program Agreement would include, but is not limited to, the proposal for the MFP the Primary Manufacturer is counteroffering and a signature by an authorized representative.
Section 1194(b)(2)(D) of the Act requires that, after receiving a counteroffer under section 1194(b)(2)(C) of the Act, CMS must respond in writing to such counteroffer. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for example, section 60.4 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, we are proposing in § 429.525(c) that we would respond in writing to a statutory written counteroffer made by the Primary Manufacturer. Although the statute does not specify a timeframe for CMS' response to the Primary Manufacturer's statutory written counteroffer, in accordance with section 1194(b)(2)(E) of the Act and as proposed in § 429.535(b), negotiations must end prior to November 1 following the selected drug publication date to avoid potential excise tax liability under 26 U.S.C. 5000D(b)(2).
As proposed in § 429.525(c)(1), in the case CMS' written initial offer is not accepted and the Primary Manufacturer submits a statutory written counteroffer, we would consider the statutory written counteroffer and either accept or reject it in writing within 30 days of receipt of the statutory written counteroffer or within 60 days of sharing the initial offer, whichever is later. When considering a statutory written counteroffer, we would evaluate whether accepting the counteroffer is consistent with the statutory directive to aim to arrive at an agreement that achieves the lowest possible MFP for the selected drug.
Section 1194(b)(2)(F) of the Act requires that CMS cannot offer a proposal for the MFP or agree to a Primary Manufacturer's proposal for the MFP for a selected drug, with respect to the initial price applicability year for the selected drug, that exceeds the ceiling determined under section 1194(c) for the selected drug and year. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for example, section 60.4 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, consistent with proposed § 429.410(a), CMS cannot accept a statutory written counteroffer from a manufacturer that exceeds the statutorily determined ceiling as defined in section 1194(c) of the Act and described in section II.E.3. of this proposed rule and in proposed § 429.410(b). Consistent with proposed § 429.440(b), CMS cannot agree to any Primary Manufacturer proposal for the MFP that is less than the Temporary Floor for Small Biotech Drugs, if applicable, as defined in section 1194(d) of the Act and proposed in section II.E.9. of this proposed rule.
Consistent with policies for implementation in section 60.4 of Negotiation Program Guidance, we propose in § 429.530(a) to provide additional price exchange opportunities through which CMS and Primary Manufacturers can initiate additional written offers and counteroffers via the CMS HPMS during the period between CMS' rejection of the Primary Manufacturer's statutory written counteroffer, if applicable, and the parties reaching an agreement on the
( printed page 36298)
MFP, or at least 8 business days before CMS issues the final offer, whichever is earlier. We believe this functionality would enable both parties to have additional flexibility to extend and consider offers and counteroffers during this time period.
We propose at § 429.530(a)(1) that the functionality for additional price exchange opportunities in the CMS HPMS would allow for the optional upload of materials which must comply with limits on the amount and format of such materials as specified by CMS, and include an optional text field to enable the offering or counteroffering party to include additional contextual information for the offer or counteroffer. As proposed in § 429.530(a)(2), only one offer or counteroffer per selected drug may be active at a time in the CMS HPMS as part of the functionality for additional price exchange opportunities. Proposed § 429.530(a)(3) states an offering/counteroffering party may revise its offer/counteroffer in the period before the other party accepts or rejects it but not afterwards. We propose at § 429.530(a)(4) that parties would not need to alternate making offers and counteroffers. Consistent with proposed § 429.410(a), CMS cannot propose an MFP or agree to any Primary Manufacturer proposal for the MFP that exceeds the statutorily determined ceiling as defined in section 1194(c) of the Act and as set forth in section II.E.3. of this proposed rule and in proposed § 429.410(b). Consistent with proposed § 429.440(b), CMS cannot propose an MFP or agree to any Primary Manufacturer proposal for the MFP that is less than the Temporary Floor for Small Biotech Drugs, if applicable, as defined in section 1194(d) of the Act and proposed in section II.E.9. of this proposed rule. Lastly, as proposed in § 429.530(a)(5), to formalize agreement on an MFP, CMS and the Primary Manufacturer both must sign an Addendum to the Negotiation Program Agreement as described in section II.C.1. of this proposed rule and in proposed § 429.200(e) that sets forth the agreed-upon MFP.
8. Notification of Final Offer and Conclusion of Negotiations (§ 429.535)
Section 1194(b)(2)(E) of the Act requires that all negotiations between CMS and the Primary Manufacturer of the selected drug shall end prior to November 1 following the selected drug publication date, with respect to the initial price applicability year. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for example, section 60.4 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, and consistent with policies for implementation as described in section 60.4 of the Negotiation Program Guidance, we are proposing in proposed § 429.535(b) that all negotiations between CMS and the Primary Manufacturer of the selected drug must end prior to November 1 following the selected drug publication date, with respect to the initial price applicability year, to avoid potential excise tax liability. As proposed in § 429.535(a), in the event neither CMS' initial offer nor the Primary Manufacturer's statutory written counteroffer were accepted, and an MFP was not agreed to during the negotiation meetings or via the additional price exchange functionality, we would send the Primary Manufacturer a “Notification of Final Maximum Fair Price Offer” and an Addendum with the final offer MFP by September 30 following the selected drug publication date defined in proposed § 429.20. This would serve as the final offer to the Primary Manufacturer for the MFP for the selected drug. This final offer would be sent only if, by September 30 following the selected drug publication date neither CMS nor the Primary Manufacturer has accepted the latest offer or counteroffer made in writing or agreed upon an MFP during the negotiation meeting process or via the additional price exchange functionality. We propose at § 429.535(a)(1), that if a final offer is sent, the Primary Manufacturer must respond in writing to this final offer by either accepting or rejecting the final offer by October 31 following the selected drug publication date.
As proposed at § 429.535(a)(2), to formalize agreement on an MFP, CMS and the Primary Manufacturer both must sign an Addendum to the Negotiation Program Agreement as described in section II.C.1. of this proposed rule and in proposed § 429.200(e) that sets forth the agreed-upon MFP. For example, when CMS prepares a written offer, we would also populate an Addendum to the Negotiation Program Agreement with the offered MFP and send that Addendum to the Negotiation Program Agreement with the written offer to the Primary Manufacturer via the CMS HPMS. If the Primary Manufacturer accepts the written offer, it would sign the Addendum to the Negotiation Program Agreement after which CMS would countersign the Addendum to the Negotiation Program Agreement.
If CMS and the Primary Manufacturer do not agree to an MFP by the deadline set forth in paragraph (b) of this section, the Primary Manufacturer would enter a period during which the excise tax may be imposed on certain sales of the selected drug. As described in 26 U.S.C. 5000D(b)(2) and 5000D(c), the Primary Manufacturer can end the period during which the excise tax may apply by agreeing to an MFP; meeting the statutory criteria for the suspension of tax; or terminating its Negotiation Program Agreement in the manner described in section II.C.2. of this proposed rule and in proposed § 429.205.
G. Renegotiation of an MFP (§§ 429.600 Through 429.620)
1. General Rule (§ 429.600)
Section 1194(f) of the Act establishes the requirements governing the identification of renegotiation-eligible drugs, the selection of drugs for renegotiation, and the renegotiation process. With respect to initial price applicability year 2028, the first year in which renegotiation could occur per section 1194(f) of the Act, we implemented policies for renegotiation in section 130 of the Negotiation Program Guidance. With respect to initial price applicability year 2029 and subsequent years, and consistent with the policies described in section 130 of the Negotiation Program Guidance, we propose to identify renegotiation-eligible drugs in accordance with section 1194(f)(2) of the Act, as described in proposed § 429.605. Next, we would select certain renegotiation-eligible drugs for renegotiation in accordance with section 1194(f)(3) of the Act, as described in proposed § 429.610. Finally, we would renegotiate MFPs for such drugs selected for renegotiation, in accordance with section 1194(f)(4) of the Act, as described in proposed § 429.620. Figure 2 depicts an overview of this proposed process.
We note that renegotiation is a component of the Negotiation Program. A Primary Manufacturer that has a Negotiation Program Agreement in effect, as discussed in proposed subpart C and section II.C. of this proposed rule, would be required to adhere to the process and deadlines described throughout this proposed rule. For example, the policies described in proposed subparts F and H (described in sections II.F. and II.H. of this proposed rule) are applicable to renegotiation unless otherwise specified herein. The
( printed page 36299)
renegotiation process would conclude with an agreed-upon MFP, unless the Primary Manufacturer chooses not to participate or chooses not to agree upon a new MFP (or CMS determines that a generic drug is approved or a biosimilar is licensed for the selected drug is subject to Bona Fide Marketing during the renegotiation period consistent with proposed § 429.130(c)(5), in which case it will no longer be subject to the renegotiation process in accordance with section 1194(f)(5) of the Act and proposed § 429.135(c)(2)). To meet their MFP effectuation obligations, Primary Manufacturers must make any agreed-upon MFP available as set forth in proposed § 429.200(b)(4).
Figure 2: Overview of Renegotiation Process Steps
If the Primary Manufacturer and CMS agree upon an MFP through the renegotiation process set forth under § 429.620, we propose at § 429.600(b)(1) that the renegotiated MFP would apply starting January 1 of the initial price applicability year for which the drug was selected for renegotiation. We also propose at § 429.600(b)(3) and consistent with the requirement in proposed § 429.200(b)(4), that to meet their MFP effectuation obligations, Primary Manufacturers of a selected drug with a renegotiated MFP must provide access to the selected drug's initial agreed-upon MFP in accordance with subpart I for all dispenses, administrations, and furnishings of the selected drug prior to such effective date for the renegotiated MFP. Additionally, we propose at § 429.600(b)(2) that the MFP that is agreed upon following renegotiation would apply to all formulations across dosage forms and strengths of the selected drug by applying the methodology set forth at proposed § 429.700. Information on MFP effectuation for 2029 and subsequent years will be included in future rulemaking.
Finally, we propose at § 429.600(c) that we would publish the list of drugs selected for renegotiation no later than the selected drug publication date (consistent with § 429.100(b)(2)).
2. Eligibility of Drugs for Renegotiation (§ 429.605)
Section 1194(f)(2) of the Act establishes the definition of a “renegotiation-eligible drug” as a selected drug for which (1) a new indication is added to the drug; (2) the drug monopoly status was not that of an extended-monopoly or a long-monopoly drug and changes to that of an extended-monopoly drug; (3) the drug monopoly status was not that of a long-monopoly drug; and changes to that of a long-monopoly drug; or (4) the Secretary determines there has been a material change to any section 1194(e)(1) or (e)(2) factor.
In accordance with section 1194(f)(1) of the Act, we propose to identify renegotiation-eligible drugs from selected drugs negotiated, or renegotiated, if applicable, with respect to prior initial price applicability years. We interpret section 1194(f)(1) of the Act to mean that the Secretary must provide for a process of renegotiation for years during the selected drug's price applicability period. For example, because calendar year 2029 will be a year within the price applicability period for drugs selected for initial price applicability years 2026, 2027, and 2028, these selected drugs may be eligible for renegotiation with respect to initial price applicability year 2029 (which would involve a renegotiation taking place in calendar year 2027) if these drugs meet any of the eligibility criteria set forth in section 1194(f)(2) of the Act and proposed § 429.605.
As a matter of operations, first, we propose in § 429.605(a) that the scope of selected drugs that would be considered for renegotiation eligibility and selection with respect to initial price applicability years beginning with initial price applicability year 2029 would include any selected drugs with an agreed-upon MFP from a prior initial price applicability year, which would include a selected drug that has an agreed-upon MFP from a prior renegotiation. A selected drug would not be subject to renegotiation if CMS determines prior to the selected drug publication date for the relevant initial price applicability year that the manufacturer of any generic drug or biosimilar, as applicable, of the selected drug is engaging in Bona Fide Marketing of such generic drug or biosimilar, based on CMS' consideration of the information set forth at proposed
( printed page 36300)
§ 429.130(a). That is, for any selected drugs with an agreed-upon MFP from a prior initial price applicability year where CMS has not yet determined that Bona Fide Marketing exists, we would proceed to review such drugs for renegotiation eligibility. If CMS determines based on consideration of the information set forth at proposed § 429.130(a) in accordance with the timing in proposed § 429.135(b)(2) and section II.B.6.d. of this proposed rule, that one or more manufacturers of an approved generic drug or licensed biosimilar, as applicable, of the selected drug, including a drug selected for renegotiation, is engaging in Bona Fide Marketing of such generic drug or biosimilar then the selected drug ceases to be subject to the renegotiation process.
Among such selected drugs, we propose to identify selected drugs which are renegotiation-eligible drugs due to a change in monopoly status as described in proposed § 429.605(b). Then, we propose to review the remaining selected drugs and identify those which are renegotiation-eligible drugs due to the addition of a new indication as described in proposed § 429.605(c) or due to a material change in any section 1194(e) factor as described in proposed § 429.605(d).
Selected drugs negotiated in initial price applicability years 2026 and 2027 were limited to those covered under Part D per section 1192(d)(2)(A) of the Act. In proposed § 429.610(a)(2), we propose to consider any selected drugs from initial price applicability years 2026 and 2027, including those with Part B utilization, for renegotiation eligibility under section 1194(f)(2) of the Act. If such drugs meet the eligibility criteria described in proposed § 429.605, they may be selected for renegotiation under section 1194(f)(3) of the Act and as described in proposed § 429.610; and may have an agreed-upon MFP after renegotiation. For such renegotiation-eligible drugs that are selected for renegotiation and for which a renegotiated MFP is agreed upon, the renegotiated MFP would apply as described in proposed § 429.600(b) and would apply to claims payable under Part B and dispenses covered under Part D, as applicable. The MFP that is agreed upon following the renegotiation process would apply to all formulations across dosage forms and strengths of the selected drug as specified in proposed § 429.600(b)(2), which would include when the selected drug is payable under Part B only, covered under Part D only, and when the drug is payable under Part B and covered under Part D. Proposed § 429.600(b)(3) would require the Primary Manufacturer of a selected drug with a renegotiated MFP to make the prior agreed-upon MFP available as set forth in proposed subpart I for all dispenses, administrations, and furnishings of the selected drug prior to the effective date of the renegotiated MFP as specified in proposed § 429.600(b)(1).
a. Selected Drugs for Which There Is a Change in Status to an Extended-Monopoly Drug
To meet the definition of a extended-monopoly drug proposed in § 429.20, the initial approval date under section 505(c) of the FD&C Act or the initial licensure date under section 351(a) of the PHS Act, as applicable, associated with the earliest-approved FDA application containing the active moiety/active ingredient (or in the case of a potential qualifying single source drug identified under § 429.125(b)(4), the distinct combination of active moieties/active ingredients) [59]
must be on or before January 1 of the year 12 years prior but no more than 16 years prior. In accordance with section 1194(f)(2)(C) of the Act, we propose in § 429.605(b)(1) that a selected drug that meets the definition of an extended-monopoly drug with respect to initial price applicability years beginning with initial price applicability year 2029, and that did not qualify as an extended-monopoly drug when the drug was selected for negotiation or a prior renegotiation would be determined to be renegotiation-eligible due to a change in status to an extended-monopoly drug.[60]
b. Selected Drugs for Which There Is a Change in Status to a Long-Monopoly Drug
To meet the definition of a long-monopoly drug, the initial approval date under section 505(c) of the FD&C Act or the initial licensure date under section 351(a) of the PHS Act, as applicable, associated with the earliest-approved FDA application containing the active moiety/active ingredient (or in the case of a potential qualifying single source drug identified under § 429.125(b)(4), the distinct combination of active moieties/active ingredients) [61]
must be on or before January 1 of the year 16 years prior. In accordance with section 1194(f)(2)(C) of the Act and the existing policy established in section 130.1.2 of the Negotiation Program Guidance, we propose at § 429.605(b)(2) that a selected drug that meets the definition of a long-monopoly drug (as proposed in § 429.20) and that did not qualify as a long-monopoly drug when the drug was selected for negotiation or a prior renegotiation would be determined to be renegotiation-eligible due to a change in status to a long-monopoly drug.
c. Selected Drugs for Which a New Indication Is Added
Section 1194(f)(2)(A) of the Act identifies a selected drug for which a new indication is added as a renegotiation-eligible drug. As described in proposed § 429.615(a), we propose to collect voluntary information submissions from Primary Manufacturers of selected drugs to inform renegotiation drug eligibility and selection.
To identify whether a new indication has been added to the FDA-approved labeling [62]
for a selected drug, we propose at § 429.605(c) to use a number of sources, for example the
Drugs@FDA database.[63]
We also propose at § 429.605(c)(2)(ii) to review voluntary submissions from Primary Manufacturers, if any, are submitted. We propose at § 429.605(c)(2)(ii)(A) that we may review off-label use for the purpose of determining whether such off-label use is a new indication for renegotiation eligibility determinations only if voluntarily submitted by the Primary Manufacturer. At § 429.605(c)(1), we propose that we would determine a drug to be renegotiation-eligible based on the addition of a new indication if the FDA-
( printed page 36301)
approved labeling has been updated to include treatment or prevention of a new disease or condition. In the renegotiation context, “new” means not considered in the previous negotiation process or renegotiation process. For example, we would not determine a selected drug to be renegotiation-eligible under section 1194(f)(2)(A) of the Act based on FDA labeling updates within a previously indicated disease or condition.
In proposed § 429.605(c)(2)(ii)(A), we propose that if the Primary Manufacturer of a selected drug submits the off-label use through the voluntary information submission process then CMS may review off-label uses when identifying new indications for renegotiation eligibility. We would apply the definition proposed at § 429.20 when reviewing an off-label use submitted by a Primary Manufacturer for consideration as a new indication for the purpose of renegotiation eligibility. Voluntary submissions from a Primary Manufacturer of an off-label use for a selected drug would not be regarded as a “new indication” for renegotiation eligibility under section 1194(f)(2)(A) of the Act if such off-label use is for a previously indicated disease or condition. For example, in the event we previously considered an off-label use during negotiation and the Primary Manufacturer later received FDA approval for that off-label use, we would not consider the new on-label use to be a new indication in accordance with section 1194(f)(2)(A) of the Act. We may consider such off-label use that is for a previously indicated disease or condition as part of the evaluation of section 1194(e) factors for material change for purposes of renegotiation eligibility under section 1194(f)(2)(D) of the Act, as described in proposed § 429.605(d).
To ensure we have appropriate time to consider applicable data, we propose at § 429.610(c)(3) that the new indication must be added to the FDA-approved labeling for the selected drug by the date of the voluntary submission as proposed at § 429.615(b). That is, the new indication must be added to the FDA-approved labeling for the selected drug on or before March 1 of the year of the selected drug publication date for the initial price applicability year for which the drug would be selected for renegotiation. The Primary Manufacturer may voluntarily submit data on a new indication, including an indication added to the FDA label or an off-label use, through the due date of the Drug Selection ICR to be specified by CMS upon approval of the Drug Selection ICR by the OMB and prior to the selected drug publication date for which the drug would be selected for renegotiation. If a Primary Manufacturer chooses to submit information on new indication, we would update the review of available indications for that selected drug, including incorporating the Primary Manufacturer's submission. We expect that there would be a low likelihood of new indications being available for selected drugs whose negotiation or renegotiation period ends on November 1 immediately prior to the Primary Manufacturer submission deadline of November 30.
d. Selected Drugs for Which There Is a Material Change in a Section 1194(e) Factor
Section 1194(f)(2)(D) of the Act directs CMS to identify a selected drug for which there has been a material change to any factor listed in section 1194(e) as a renegotiation-eligible drug and provides CMS with the discretion to determine what constitutes a “material change.”
We propose to consider a change(s) to a section 1194(e) factor for a selected drug to be material if the change(s) to the factor would reasonably be expected to meaningfully alter our consideration of that factor within the context of renegotiation offers and counteroffers, including the initial offer as determined in proposed subpart F, as compared with our consideration of that factor within the context of offers, including the initial offer and, if applicable, counteroffers, during the most recent prior negotiation or renegotiation process for the selected drug. For purposes of determining whether a selected drug is renegotiation-eligible under section 1194(f)(2)(D) of the Act, we would evaluate available information pertaining to section 1194(e) factors (as listed in proposed § 429.505(b) and (d) and section II.F.2.b. of this proposed rule) to determine if there is a material change. We also propose to consider voluntary submissions by a Primary Manufacturer of the information discussed in proposed § 429.615(a) to inform our determination.
This approach complements our review of new indications described in § 429.605(c) and discussed in section II.G.2.c. of this proposed rule as we would consider prescribing information (see section 1194(e)(2)(B) of the Act) as part of this material change evaluation. As part of that inquiry, we would review the FDA-approved labeling for a material change in prescribing information as described in proposed § 429.605(d)(1) and, in doing so, would also capture the impact of labeling updates within a previously indicated disease or condition during such review (for example, expansion of an existing indication to include an additional age group(s)). That is, labeling updates within a previously indicated disease or condition would not be regarded as a “new indication” for renegotiation eligibility under section 1194(f)(2)(A) of the Act but would be considered as part of the review of section 1194(e) factors for material change for purposes of renegotiation eligibility under section 1194(f)(2)(D) of the Act.
To provide sufficient time to consider the applicable information to determine whether a change in a factor listed in section 1194(e) of the Act is material, we would consider the applicable information that is available on or before September 30 of the calendar year before the selected drug publication date for which the drug would be selected for renegotiation. Our review by September 30 would not be inclusive of information submitted by the Primary Manufacturer through the voluntary submission due November 30. For example, this may include information on any offers or counteroffers made on or before September 30 of the calendar year before the selected drug publication date for which the drug would be selected for renegotiation, including MFPs that were agreed upon for the selected drug or its therapeutic alternatives. Should the Primary Manufacturer choose to submit data through the voluntary submission described in proposed § 429.615(a), then we may update our review of material change(s) with new applicable information.
Table 2 provides a few illustrative examples of this approach to determining a material change(s) to a section 1194(e) factor for the purpose of determining renegotiation eligibility. We reiterate that these are illustrative examples and the potential results may vary in all cases depending on the fact patterns of a given drug.
( printed page 36302)
Table 2—Illustrative Example Scenarios and Potential Result in Determination of Material Change
Example
scenario
Potential
result for changes in section
1194(e)
factor
New clinical data is released showing increased clinical value for a greater number or new group of individuals who are prescribed a selected drug
CMS may determine that in this scenario that this new data would likely meaningfully impact CMS' consideration of section 1194(e)(2)(C) of the Act in the context of offers and counteroffers. Therefore, this change may be considered a material change.
Unit cost of production and distribution increases from $1.00 per unit to $1.50 per unit
CMS may determine that in this scenario this change would not likely meaningfully impact CMS' consideration of section 1194(e)(1)(B) of the Act in the context of offers and counteroffers. Therefore, this change alone may not be considered a material change.
A therapeutic alternative is now generic, the price for the therapeutic alternative drops significantly, and utilization of the therapeutic alternative increases significantly
CMS may determine that in this scenario this new data would likely meaningfully impact CMS' consideration of section 1194(e)(2)(A) of the Act in the context of offers and counteroffers. Therefore, this change may be considered a material change.
New clinical data is released showing increased clinical efficacy of the selected drug for one of its indications; a new black box warning indicates additional safety concerns for the selected drug, limiting its use
CMS may determine that in this scenario the net impact of these changes would not meaningfully impact CMS' consideration of section 1194(e)(2)(C) of the Act in the context of offers and counteroffers. Therefore, these changes alone may not be considered a material change.
An indication for a selected drug was not covered under Part D or payable under Part B at the time the drug was previously negotiated. The indication is now covered under Part D and/or payable under Part B and there are no alternative treatments available for that indication
CMS may determine that in this scenario the newly covered indication for which there are no alternative treatments to the selected drug would meaningfully impact CMS' consideration of section 1194(e)(2)(D) of the Act in the context of offers and counteroffers. Therefore, this change may be considered a material change.
3. Selection of Drugs for Renegotiation (§ 429.610)
Section 1194(f)(3) of the Act directs CMS to select drugs for renegotiation from the identified renegotiation-eligible drugs (described in proposed § 429.605). Sections 1194(f)(3)(A) and (B) of the Act require all such drugs eligible for renegotiation due to a change in monopoly status to either an extended-monopoly drug or a long-monopoly drug, as specified in proposed § 429.610(b), be selected for renegotiation. Section 1194(f)(3)(C) of the Act states that among the remaining renegotiation-eligible drugs (that is, selected drugs that are determined to be renegotiation-eligible due to a new indication or a material change in a section 1194(e) factor), CMS shall select renegotiation-eligible drugs for which CMS expects renegotiation is likely to result in a significant change in the previously agreed-upon MFP. Section 1194(f)(3)(C) of the Act provides CMS with the discretion to make determinations on when a renegotiation is “likely to result in a significant change” in the MFP.
a. Selecting Drugs for Renegotiation Among Renegotiation-Eligible Drugs due to a Change in Monopoly Status
In accordance with section 1194(f)(3)(A) and (B) of the Act and consistent with the existing policies established in section 130.2 of the Negotiation Program Guidance, we propose at § 429.610(a) to select for renegotiation all drugs that are determined to be renegotiation-eligible due to a change in monopoly status to either an extended-monopoly drug or a long-monopoly drug as set forth in proposed § 429.605(b).
b. Selecting Drugs for Renegotiation Among Renegotiation-Eligible Drugs due to a New Indication or a Material Change in a Section 1194(e) Factor
In accordance with section 1194(f)(3)(C) of the Act and consistent with the existing policies established in section 130.2.1 of the Negotiation Program Guidance, we propose to select remaining renegotiation-eligible drugs (that is, selected drugs that are determined to be renegotiation-eligible due to a new indication or a material change in a section 1194(e) factor), for renegotiation if we expect renegotiation is likely to result in a significant change in the MFP.
To inform our determination of whether renegotiation is likely to result in a significant change in the MFP, we propose to consider two criteria to effectuate this statutory standard, as discussed in greater detail in this section. We propose that the evaluation of these two criteria would be a holistic inquiry based on the totality of the information available and the circumstances of the remaining renegotiation-eligible drug(s). The first criterion that we propose in § 429.610(b)(1)(i) is that we would consider whether a new indication(s) or material change(s) would be likely to result in a renegotiated MFP that represents a 15 percent or greater change relative to the current MFP upon engaging in renegotiation with the Primary Manufacturer. The second proposed criterion in § 429.610(b)(1)(ii) is to consider whether such a change in the MFP for the remaining renegotiation-eligible drug(s) would have a significant impact on the Medicare Program. We believe these criteria are important to consider holistically to understand the circumstances for each selected drug.
Our evaluation for these two criteria would be a holistic inquiry based on the totality of the information available and the circumstances of the remaining renegotiation-eligible drug(s), including nonfinancial information, such as the expiration of an exclusivity period under the FD&C Act or the PHS Act, and would support our determination of whether renegotiation is likely to result in a significant change, whether an increase or decrease, to the MFP for the remaining renegotiation-eligible drug(s). The scope of information considered may extend beyond the scope of information evaluated for renegotiation
( printed page 36303)
eligibility to include a CMS-led review of the information sources discussed in proposed § 429.510(b) (described in section II.F.2. of this proposed rule) and the evaluation of such sources as discussed in proposed § 429.510(c), (e), and (f) (described in section II.F.3.d. of this proposed rule) pertaining to section 1194(e)(1) and section 1194(e)(2) factors. We propose to consider these information sources and others to inform whether a renegotiation-eligible drug would be selected for renegotiation because these sources may inform a potential future renegotiation process, including the potential development of the initial offer and subsequent offers, as well as our consideration of potential counteroffers.
We propose that when a remaining renegotiation-eligible drug is subject to this evaluation, we will only select such drug for renegotiation if that renegotiation-eligible drug meets both of these criteria. We believe each of these criteria are of equal importance when considering whether renegotiation is likely to result in a significant change in the MFP otherwise negotiated. We believe that when considering drug selection for renegotiation from among renegotiation-eligible drugs, described previously, the evaluation of the proposed criteria would indicate that renegotiation is both likely to change the MFP and that the change in MFP would be significant, in accordance with the directive under section 1194(f)(3)(C) of the Act to select remaining renegotiation-eligible drugs when CMS “expects renegotiation is likely to result in a significant change in the maximum fair price otherwise negotiated.” We interpret section 1194(f)(3)(C) of the Act to require that for a drug that is eligible for renegotiation based on a new indication or material change to a factor listed at section 1194(e) of the Act, we must expect that renegotiation would likely result in a change to the MFP, but also that such change must be considered significant, such that the magnitude of change is meaningful and that such change would be significant for CMS. We believe that evaluating the proposed criteria provides transparency into how we intend to implement section 1194(f)(3)(C) of the Act. As a matter of process and for additional clarity, if a remaining renegotiation-eligible drug(s) were to fail to meet either criterion, we would not proceed to examine the other criterion given that a remaining renegotiation-eligible drug(s) must meet both criteria to be selected for renegotiation.
In establishing the first criterion (the likelihood that the new indication or material change would result in a renegotiated MFP that would likely represent a 15 percent or greater change relative to the current MFP) we reviewed sections 1194(c)(3)(A) through (C) of the Act, which provide the applicable percentage of non-FAMP used to establish a ceiling during negotiation. The applicable percentages are 75 percent for short-monopoly drugs and vaccines, 65 percent for extended-monopoly drugs, and 40 percent for long-monopoly drugs (as described further in proposed § 429.410). Given that, under sections 1194(c)(3)(A) through (C) of the Act, a change in monopoly status is associated with a percent reduction in the ceiling for negotiation and that such change in monopoly status of a selected drug results in eligibility and selection for renegotiation per sections 1194(f)(2)(B) and (C) of the Act and sections 1194(f)(3)(A) and (B) of the Act, we believe this range of percent change is informative for interpreting what the term “significant change” means in the context of the statute.
We calculated the percent change in the applicable percentage for a drug that had a change in monopoly status from a short-monopoly drug (75 percent) to a long-monopoly drug (40 percent), which is approximately 46.7 percent change; this represents the maximum percent change in the applicable percentage that would correspond with a selected drug becoming renegotiation-eligible and selected for renegotiation. We also calculated the percent change in the applicable percentage for a drug that had a change in monopoly status from a short-monopoly drug (75 percent) to an extended-monopoly drug (65 percent), which is approximately 13.3 percent change; this represents the minimum percent change in the applicable percentage that would correspond with a selected drug becoming renegotiation-eligible and selected for renegotiation. We rounded these percent changes to the nearest 5 percent, resulting in a range of percent change from 15 to 45 percent that we believe represent an informative range for interpreting what a “significant change” means in the context of the statute, as discussed previously. We believe that a potential change in the MFP between 15 and 45 percent would be significant for the Medicare program and Medicare beneficiaries. We propose to use minimum percent change that corresponds with a selected drug becoming renegotiation-eligible and selected for renegotiation, that is 15 percent change, so as to not preclude the benefits that could come from a change in the MFP that is at least 15 percent. The agency also considered the resources invested in renegotiation. We believe that the resources invested for both CMS and the Primary Manufacturer in a renegotiation process that would likely result in a change to the MFP of 15 percent or greater is an appropriate investment for the potential resulting change.
We also considered the option of evaluating the likelihood that a new indication or material change would result in a 35 percent or greater change to the MFP. In connection with the public comment period for the Negotiation Program Guidance for initial price applicability year 2028 a few commenters noted that the difference between the applicable percentage for short-monopoly drugs and vaccines (75 percent) and long-monopoly drugs (40 percent) is 35 percent and therefore they recommended we adopt an expected change to the MFP of 35 percent as the first criterion indicating a potential change to the MFP is significant. We note that a 35 percent change would fall in the range calculated based on the percent change in the applicable percentages as described previously. However, we believe using the minimum percent change in the applicable percentage that corresponds with a selected drug becoming renegotiation-eligible and selected for renegotiation provides CMS with the best opportunity to both clearly define what constitutes a “significant change to the MFP” and to meet our statutory obligation per section 1194(b)(1) of the Act to establish a process that aims to achieve the lowest MFP for each selected drug. Additionally, we believe using a range based on the
percent change
in the applicable percentage is more aligned with section 1194(f)(3)(C) of the Act that specifies that CMS should select drugs for renegotiation that are likely to have a significant
change
in the MFP [emphasis added]. The suggested approach from commenters represents looking at the arithmetic difference in applicable percentages, rather than the change in applicable percentages. Use of percent change rather than the difference in the applicable percentages has practical importance in that our language more closely aligns with statute, meaning that a renegotiation would result in a change in the MFP. Given the variance in expected percent change in the MFP, a numerical value of 15 percent establishes the minimum amount that may have a meaningful effect for beneficiaries and the Medicare program
( printed page 36304)
and allows CMS to potentially consider more renegotiation-eligible drugs for renegotiation selection, whereas a higher number, like 35 percent, may normalize that any change less than 35 does not have a meaningful effect and may result in the selection of fewer renegotiation-eligible drugs for renegotiation. A 15 percent or greater change is consistent with the range of percent reductions in the ceiling that is statutorily defined for drugs eligible and selected for renegotiation due to monopoly status changes.
We also believe this criterion serves to promote transparency and consistency in the approach to selecting drugs for renegotiation in accordance with section 1194(f)(3)(C) of the Act. Therefore, in an effort to promote transparency and consistency, we propose to include this criterion of considering the likelihood that the new indication(s) and/or material change(s) to a section 1194(e) factor would result in a renegotiated MFP that would represent a 15 percent or greater change relative to the current MFP, which can help provide clarity for interested parties and the public on when we may select remaining renegotiation-eligible drugs in accordance with section 1194(f)(3)(C) of the Act.
We recognize that there may be other circumstances related to the renegotiation-eligible drug that may make selecting the drug for renegotiation less reasonable, for example the impending entry of a generic or biosimilar to market. As such, we propose the complementary criterion of evaluating whether such a change would have a significant impact to the Medicare program. For the second proposed criterion for determining whether renegotiation is likely to result in a significant change in the MFP, we propose at § 429.610(b)(1)(ii) to consider the impact on the Medicare program of such a change in the renegotiated MFP. For example, if we determine there was a likelihood that the new indication(s) or material change(s) could result in a renegotiated MFP that represents a 15 percent or greater change relative to the current MFP, we would consider the financial impact to the Medicare program and Medicare beneficiaries by reviewing associated changes in expenditures and beneficiary cost-sharing. In doing so, we seek to incorporate consideration of whether such change in MFP warrants the time and resource investment by the Primary Manufacturers and CMS in the renegotiation process. As an example, consider a scenario where a selected drug becomes renegotiation-eligible and there was a likelihood that the percentage change in the MFP would be greater than 15 percent. However, in this example scenario, patents on the selected drug will have expired before a renegotiated MFP would take effect, and we determine that this patent expiration will likely result in the introduction of robust generic competition. In this example scenario, we believe that: (1) it is likely there will be a substantial price drop in the market price for the selected drug unrelated to a renegotiated MFP; and (2) the drug will no longer be a selected drug on January 1 of the initial price applicability year because such drug has an approved generic drug that is subject to Bona Fide Marketing. As such, we may not select this hypothetical selected drug for renegotiation since the impact to the Medicare program might be minimal and does not warrant the considerable investment of time and resources by the Primary Manufacturer or CMS.
We propose to consider the totality of other available information to determine whether renegotiation is likely to result in a change in MFP of 15 percent or greater and that such a change would have a significant impact on the Medicare program. This would include an initial evaluation of the evidence available for a renegotiation-eligible drug. This holistic consideration of the available information and circumstances for renegotiation drug selection provides for the consideration of drug- and fact-specific circumstances. This approach would also maintain consistency with the process for developing the initial offer, which considers the totality of available evidence as described in proposed §§ 429.510 and 429.620(f).
In making this proposed determination, we do not presume that the result of a renegotiation would reflect these estimations, rather, these criteria would serve to support our selection determination in alignment with the requirements set forth in section 1194(f)(3)(C) of the Act and to help provide clarity for interested parties and the public. We note that no criteria to select drugs for renegotiation can predict the actual outcome of a renegotiation. A determination by CMS that renegotiation is likely to result in a significant change in MFP does not restrict the possibilities for the outcome of renegotiation. Any given renegotiation, informed by data on section 1194(e) factors available during the renegotiation period, could result in an increase in MFP, decrease in MFP, or no change in MFP. Further, the magnitude of the change in MFP could be higher or lower than 15 percent. Similarly, we do not presume that the result of a renegotiation will reflect these approximations, for example, with respect to the impact of an agreed upon MFP following the renegotiation process on the Medicare program.
We considered reviewing the remaining renegotiation-eligible drugs solely based on the criterion related to the impact on the Medicare program. We believe this criterion is an important component of defining, as a procedural matter, what constitutes a “significant change to the MFP otherwise negotiated”, that is, we believe it is necessary when paired with some expectation of a change in the actual MFP but not sufficient to define a “significant change”.
We also acknowledge that for renegotiation eligibility and selection of selected drugs that are negotiated or renegotiated for the initial price applicability years 2 years prior and immediately before the initial price applicability year for which eligibility and selection for renegotiation is being conducted, the time between agreeing upon an MFP (through negotiation or renegotiation), and the review for renegotiation eligibility for the applicable initial price applicability year may be relatively short. The relatively short period between the negotiation or renegotiation of an MFP and our subsequent review of such drug for renegotiation may make it less likely that recently negotiated or renegotiated drugs would meet the material change criteria proposed at § 429.610(b) and described in section II.G.3.b. of this proposed rule.
Table 3 provides illustrative examples of how we may consider these criteria to determine whether a renegotiation-eligible drug would be selected for renegotiation.
( printed page 36305)
Table 3—Illustrative Example Scenarios and Potential Result for Selection of Renegotiation-Eligible Drugs Due to a New Indication or Change in a Section 1194(
e
) Factor
Example scenario
Potential result
New comparative clinical effectiveness data has become available that is favorable for the selected drug compared to therapeutic alternatives (for example, studies are published showing the selected drug has a greater positive effect on clinical outcomes relative to therapeutic alternatives), but the ceiling* represents a <15% increase in the MFP
It is not possible for renegotiation to result in a 15% increase in the MFP (because the ceiling* is less than 15% higher than current MFP), so the selected drug would not be selected for renegotiation.
New comparative clinical effectiveness data has become available that is unfavorable for the selected drug compared to its therapeutic alternatives (for example, studies are published showing the therapeutic alternative has a greater positive effect on clinical outcomes relative to the selected drug) and the MFP is much higher than competitors' prices. If such data were used during renegotiation with the Primary Manufacturer, the renegotiated MFP would likely decrease by 15% or more compared to the original MFP
The selected drug would be selected for renegotiation if consideration of the other criterion supports the determination that renegotiation is likely to result in a significant change to the MFP.
New comparative clinical effectiveness data has become available that is favorable for the selected drug compared to therapeutic alternatives (for example, studies are published showing the selected drug has a greater positive effect on clinical outcomes relative to the therapeutic alternatives, and the ceiling represents a >15% increase in the MFP.) Upon renegotiating with the Primary Manufacturer, the renegotiated MFP would likely increase by 15% or more compared to the original MFP
* See proposed § 429.620(b) for additional detail on the ceiling that would be used for renegotiation.
4. Data Collection To Inform Renegotiation Eligibility, Selection, and Renegotiation of the MFP for a Selected Drug (§ 429.615)
Section 1194(f)(1) of the Act directs CMS to provide for a process to renegotiate the MFP for selected drugs that are determined to be renegotiation-eligible under section 1194(f)(2) of the Act and selected in accordance with section 1194(f)(3) of the Act. With respect to initial price applicability year 2028, we implemented these requirements through guidance, including section 130.3 of the Negotiation Program Guidance. With respect to initial price applicability year 2029 and subsequent years, consistent with the policies for implementation as described in section 130.3 of Negotiation Program Guidance, we propose two data collections in § 429.615 for purposes of informing renegotiation eligibility, selection, and renegotiation of the MFP for a selected drug. The first data collection as described in § 429.615(a) would inform renegotiation eligibility and selection; the second data collection as described in § 429.615(b) would inform the renegotiation process for drugs selected for renegotiation. We will issue two ICRs, each for a 60-day public comment period, alongside this proposed rule. The ICRs include more details regarding how manufacturers can submit data, including the format for data submission. The associated burdens for these ICRs are also discussed in section IV. of this proposed rule.
a. Voluntary Information Submission From Primary Manufacturers To Inform Renegotiation Eligibility and Selection for Selected Drugs
Sections 1194(f)(2) and 1194(f)(3) of the Act do not identify any specific source for the information used to inform renegotiation eligibility and selection. We believe that the information necessary to determine renegotiation eligibility and selection for drugs with an agreed upon MFP is available without submission by the Primary Manufacturer or other interested parties. The sources of information we intend to use for renegotiation eligibility are listed in proposed § 429.605(c)(2), (d)(2), and (d)(3) and further discussed in sections II.G.2.c. and II.G.2.d. of this proposed rule. The sources of information we intend to use to select drugs for renegotiation from among those that are determined to be renegotiation-eligible are listed in proposed § 429.610(b)(2) and further discussed in section II.G.3.b. of this proposed rule. However, we believe input from the Primary Manufacturer on new indications, including off-label uses, and material changes to any factor listed at section 1194(e) of the Act, if applicable, could provide additional information or further validate- our review. To minimize burden on the Primary Manufacturer, we propose to make this data submission voluntary. We do not intend to review information submitted by a Primary Manufacturer if its selected drug is renegotiation-eligible due to a change in monopoly status as described at proposed § 429.605(b) as sections 1194(f)(3)(A) and (B) of the Act require CMS to select all drugs with a change in monopoly status described at proposed § 429.605(b) regardless of whether the drug has a new indication or a material change to a factor listed at section 1194(e) of the Act; no additional information would be required to make such a determination. We may consider the Primary Manufacturer's past data submissions, including any updates to such information, to inform renegotiation eligibility and selection, and to use such information during renegotiation if a drug is selected for renegotiation.
We also propose at § 429.605(c)(2)(ii)(A) that we would only review off-label uses as a new indication for the purpose of determining renegotiation eligibility if the off-label use meets the definition proposed in § 429.20 and is submitted by the Primary Manufacturer in this voluntary submission. We believe that the review of off-label use is discretionary and not explicitly mandated in section 1194(f)(2)(A) of the Act. Further, there is no single reliable directory or source of information regarding off-label use of FDA-approved drugs. Therefore, consideration of off-label use would be based on the evidence available for a given selected drug. Should a Primary Manufacturer of a selected drug decide to submit information about off-label use of the selected drug through this process, the
( printed page 36306)
Primary Manufacturer should indicate in their submission if the referenced off-label use meets the definition of off-label use as proposed in § 429.20.
Consistent with the policies for implementation provided in section 130.3.1 of the Negotiation Program Guidance, we propose at § 429.615(a) to provide each Primary Manufacturer of a selected drug the opportunity to submit information on new indications, including new off-label use, and new or updated information on the factors listed in section 1194(e) of the Act to inform renegotiation eligibility (as described in proposed § 429.605(c) and (d)) and selection (as described in proposed § 429.610(b)). We intend to collect this information through the Drug Selection ICR (CMS-10844, OMB 0938-1443) which would include revisions to collect such information to inform renegotiation eligibility and selection. We would deem this voluntary submission to be proprietary information from the Primary Manufacturer that is protected from disclosure in accordance with the confidentiality policies described in section II.D.1. of this proposed rule and in proposed § 429.300.
b. Data Collection From Primary Manufacturers and Other Interested Parties for Renegotiation of the MFP
In accordance with a Primary Manufacturer's responsibility under section 1193(a)(5) of the Act and under the Negotiation Program Agreement (set forth in proposed § 429.200(b)) and consistent with the policies for implementation provided in section 130.3.2 of Negotiation Program Guidance, we solicit comment on these proposed policies for data collection in the context of renegotiation of the MFP, including whether it may be preferable to more expressly establish CMS' intent to align with data collection in the context of the original negotiation process by replacing the text in proposed § 429.615(b) with text that parallels that used in § 429.505(a).
Specifically, we propose at § 429.615(b)(1)(i) that the Primary Manufacturer of a drug selected for renegotiation would be required to submit information regarding the section 1194(e)(1) factors for the selected drug to CMS, inclusive of NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer, as occurs following selection for negotiation. We also propose at § 429.615(b)(1)(ii) that if the drug selected for renegotiation was originally selected for negotiation for initial price applicability year 2026 or 2027 and has not previously been selected for renegotiation for initial price applicability year 2028 or thereafter, the Primary Manufacturer would be required to submit, if available, non-FAMP, unit type, and total unit volume for all NDC-11s of the selected drug payable under Part B, not covered under Part D, and for which the Primary Manufacturer did not report such information with the Primary Manufacturer's data submission for the initial price applicability year for which the selected drug was first selected for negotiation, for the same calendar years for which non-FAMP data was reported in the Primary Manufacturer's data submission for the initial price applicability year for which the selected drug was first selected for negotiation. Such data, if applicable, will be used to update the non-FAMP used in the renegotiation ceiling calculation described at proposed § 429.620(b) and further discussed in section II.G.5.a. of this proposed rule. The submission of information described at proposed § 429.615(b)(1) would be due at the same time the submission described at § 429.505(a) is due for drugs selected for negotiation for the same initial price applicability year, that is by 11:59 p.m. PST on March 1 of the year of the selected drug publication date for the initial price applicability year for which the drug was selected for negotiation or renegotiation, as applicable. This information would be submitted through the Drug Price Negotiation ICR (CMS-10849, OMB 0938-1452).
We also propose at § 429.615(b)(2) to provide any interested party with the opportunity to submit information related to section 1194(e)(2) factors, as occurs for negotiation. We propose at § 429.615(b) that the submission of section 1194(e)(2) information would be due at the same time such information is due for drugs selected for negotiation for the same initial price applicability year, that is March 1 of the year of the selected drug publication date for the initial price applicability year for which the drug is selected for renegotiation. We intend to collect this information through the Drug Price Negotiation ICR (CMS-10849, OMB 0938-1452) which will include revisions to collect the mandatory and optional information regarding the section 1194(e)(2) factors (set forth in proposed in § 429.505(d)(3)) to inform renegotiation of selected drugs). To minimize burden on Primary Manufacturers, we propose that this information collection be a streamlined version of that used for negotiation.
5. Renegotiation Process (§ 429.620)
In accordance with section 1194(f)(4)(B) of the Act, we intend, to the extent practicable, for the renegotiation process to be consistent with the methodology and process established under section 1194(b) of the Act. We propose to implement policies consistent with the policies for implementation set forth in section 130.4 of the Negotiation Program Guidance, with revisions discussed later in this section based on implementation experience.
a. Determining the Ceiling
Section 1194(f)(4)(B) of the Act requires that the process specified for renegotiation must, to the extent practicable, be consistent with the methodology and process established under section 1194(b) of the Act and in accordance with sections 1194(c), (d), and (e) of the Act. Section 1194(f)(4)(B) of the Act further provides that for purposes of applying sections 1194(c)(1)(A) and 1194(d) of the Act, the reference to the first initial price applicability year of the price applicability period with respect to such drug shall be treated as the first initial price applicability year of such period for which the MFP established pursuant to such renegotiation applies, including for applying section 1194(c)(3)(B) of the Act in the case of renegotiation-eligible drugs described in section 1194(c)(3)(A) of the Act and section 1194(c)(3)(C) of the Act in the case of renegotiation-eligible drugs described in section 1194(c)(3)(B) of the Act.
Consistent with section 1194(b)(2)(F)(i) of the Act, in renegotiating the MFP of a selected drug, we propose at § 429.620(b) that CMS would not make an offer (or agree to a counteroffer) for an MFP that exceeds the ceiling determined under section 1194(c) of the Act, as adjusted, if applicable, as described in this section II.G.5.a. of this proposed rule. Additionally, consistent with the policies for implementation set forth in section 130.4.1 of the Negotiation Program Guidance, we propose at § 429.620(a) to use the same methodology to calculate the ceiling amounts under section 1194(c) of the Act for renegotiation as is used for negotiation (described in proposed § 429.410) with limited revisions to reflect the updates required by statute.
For the purposes of calculating the ceiling amount under section 1194(c) of the Act, we interpret section 1194(f)(4)(B) of the Act as permitting limited updates to the ceiling applied during negotiation when a drug is first
( printed page 36307)
selected. We interpret section 1194(f)(4)(B) of the Act as directing CMS to update the applicable percents applied to selected drugs determined to be renegotiation-eligible due to a change in monopoly status under sections 1194(f)(3)(A) and 1194(f)(3)(C) of the Act, in accordance with the applicable percent for such monopoly statuses described in sections 1194(c)(3)(B) and 1194(c)(3)(C) of the Act. In contrast, section 1194(f)(4)(B) of the Act does not specify that the agency should otherwise recalculate the ceiling amounts described under sections 1194(c)(1)(B) and 1194(c)(1)(C) of the Act, including any redeterminations of the time periods used to calculate such amounts. We interpret the absence of such language in statute, in the context of the more specific instructions to update the applicable percents applied with respect to certain renegotiation-eligible drugs, to provide that CMS largely may not recalculate the ceiling amounts determined with respect to the negotiation for which the drug was first selected. We believe that a contrary interpretation, under which we recalculate the ceiling amounts described under sections 1194(c)(1)(B) and 1194(c)(1)(C) of the Act using the most recent data available with respect to the initial price applicability year for which a drug is selected for renegotiation, would likely result in a renegotiation ceiling that, over time, exclusively decreases. Such an interpretation would effectively preclude the possibility of renegotiating a higher MFP for the selected drug, and such a limitation is not set forth in the statute, including in the criteria for renegotiation selection described under section 1194(f)(3)(C) of the Act. We do not believe such an interpretation is the best reading of the statutory text set forth in section 1194(f) of the Act.
In accordance with this interpretation, and consistent with the policies for implementation set forth in section 130.4.1 of the Negotiation Program Guidance, we propose to determine the ceiling applicable to renegotiation using the ceiling amounts determined with respect to the negotiation for which a drug is first selected, with limited updates discussed below, including to account for the statutory directive to update the applicable percent for certain renegotiation-eligible drugs.
We propose at § 429.620(b)(1) to update the ceiling amounts under section 1194(c) of the Act to incorporate NDC-11s that are payable under Part B if a drug that was originally selected for initial price applicability year 2026 or 2027 is selected for renegotiation and has such NDC-11s. In accordance with section 1192(d)(1) of the Act, initial price applicability years 2026 and 2027 were the only years in which CMS was directed to limit drug selection to drugs covered under Part D and as such, the ceiling amount under section 1194(c) of the Act did not include NDC-11s payable under Part B, if any, since such NDCs were not considered negotiation-eligible at that time. This proposal would address the fact that the Negotiation Program is no longer limited to drugs covered under Part D. Because renegotiation under this proposed rule applies to initial price applicability year 2029 and beyond, any NDC-11s of a selected drug that are on the HCPCS-NDC crosswalks would be included in the negotiation process, including for renegotiation, and thus any such NDC-11s should be incorporated into the ceiling used for such renegotiation. Not doing so would mean applying a ceiling amount under section 1194(c) of the Act that is based only on NDC-11s covered under Part D of the selected drug, despite the renegotiation process and the MFP that may result from such renegotiation applying to all NDC-11s covered under Part D and/or payable under Part B.
We propose at § 429.620(b)(2) to update the applicable percent under section 1194(c)(3)(C) of the Act for purposes of calculating the non-FAMP ceiling under section 1194(c)(1)(C) of the Act for drugs selected for renegotiation due to a change in monopoly status. Consistent with § 429.435(a)(4)(vi), the applicable percent would be based on the initial approval date associated with the earliest-approved FDA application belonging to the NDA holder or BLA holder, as described in section II.B.6. of this proposed rule, for the selected drug and the initial price applicability year for which the drug is selected for renegotiation.
Finally, for all drugs selected for renegotiation, we propose at § 429.620(b)(3) to adjust the amounts considered for the ceiling described under section 1194(c) of the Act and proposed at § 429.410 by the percent increase in the CPI-U from July of the calendar year that is 2 years prior to the initial price applicability year of the most recent agreed upon MFP through July of the calendar year prior to the calendar year in which the drug is selected for renegotiation. This approach is consistent with sections 1194(f)(4) and 1195(b)(1)(A) of the Act and proposed § 429.705 under which we would publish an updated MFP increased by the annual percentage increase in the CPI-U. That is, for each year subsequent to the first initial price applicability year of the price applicability period for the selected drug with an agreed-upon MFP, the annual percentage increase in the CPI-U would be based on the 12-month period ending with the July immediately preceding November 30 of the year that is 2 years prior to such subsequent year. For example, if a drug was selected for negotiation for initial price applicability year 2026, CMS and the Primary Manufacturer agreed upon an MFP, and the drug was selected for renegotiation for initial price applicability year 2029, we would increase the amounts considered for the ceiling that were applicable to the initial price applicability year 2026 negotiation (incorporating any Part B data as applicable) by the increase in CPI-U from July 2024 through July 2027. This increase represents the cumulative inflation adjustment that we would have applied to the initial price applicability year 2026 MFP through the most recent published MFP files at the time that the drug is selected for renegotiation (which would be the files for MFPs effective January 1, 2029, published in November 2026). If we did not similarly increase the amounts considered for the ceiling by the same inflation adjustment that we apply to the MFP under section 1195(b)(1)(A) of the Act, the inflation-adjusted MFP could, over time, approach and eventually exceed the original ceiling amount. This could preclude our ability to select a drug for renegotiation and renegotiate the MFP for such drug that may otherwise be renegotiation-eligible and selected in accordance with sections 1194(f)(2) and 1194(f)(3) of the Act and under the criteria proposed in §§ 429.605 and 429.610, respectively. This potential limitation on CMS' ability to renegotiate increased MFPs would be inconsistent with section 1194(f)(3)(C) of the Act, which does not limit selection for renegotiation to instances exclusively in which renegotiation would likely result in a decrease in the MFP and thus contemplates that renegotiation may be appropriate where we expect renegotiation would be likely to result in an increased MFP.
b. Negotiation Factors
Section 1194(f)(4)(B) of the Act requires that the renegotiation process shall be consistent with the methodology and process for negotiation, to the extent practicable. As such, we propose at § 429.620(c) to consider the negotiation factors listed at sections 1194(e)(1) and (e)(2) of the Act inclusive of information submitted or shared about the factors listed at sections 1194(e)(1) and (e)(2) of the Act
( printed page 36308)
in any prior negotiation or renegotiation(s) as described in proposed § 429.505 and discussed in II.F.2. of this proposed rule consistent with the policies for implementation at section 130.4.2 of Negotiation Program Guidance.
c. Methodology for Developing an Initial Offer
Consistent with policies for implementation as described in section 130.4.2 of Negotiation Program Guidance, we propose at § 429.620(d) that the methodology for developing the initial offer for all drugs selected for renegotiation be the same process and timeline set forth for development of the initial offer for the negotiation process under proposed § 429.510. This would apply the process described in proposed § 429.510 including the review of information related to the section 1194(e)(1) and 1194(e)(2) factors described in proposed § 429.505. By uniformly adopting the initial offer process described in proposed § 429.510 for the purpose of the development of all initial offers in the context of renegotiation this aspect of renegotiation process would be fully aligned with the negotiation process consistent with the requirement at section 1194(f)(4)(B) of the Act. We note that we would apply the ceiling calculated for renegotiation per proposed § 429.615(b) when adopting the process described in proposed § 429.510 where applicable. As part of this proposed policy, we intend to require the Primary Manufacturer to submit the most recent agreed upon MFP as part of the data submission requirement described in proposed § 429.615(b)(1) through the Drug Price Negotiation ICR (OMB 0938-1452) discussed in section II.G.4.b. of this proposed rule. We believe that considering the MFP during the development of the initial offer would better allow us to incorporate the impact of the offer and counteroffer process from the prior negotiation or renegotiation into the initial offer for the current renegotiation.
Submission of the MFP would be a new collection requirement within the existing data submission. All drugs selected for renegotiation will have an agreed upon MFP and adding collection of the MFP as a data point in this submission requirement would be standard for all such selected drugs. We believe the MFP is an important data point to consider as part of our consideration of “market data and revenue and sales volume data for the drug in the United States” (the factor listed at section 1194(e)(1)(E) of the Act) as it is a Medicare-specific price that, per section 1193(a) of the Act, Primary Manufacturers are required to make available to MFP-eligible individuals (as defined at section 1191(c)(2) of the Act). We also believe that considering the MFP under the factor listed at section 1194(e)(1)(E) of the Act and therefore as a part of the totality of data submitted related to section 1194(e)(1) factors is consistent with the process used to develop the initial offer for negotiation as described at proposed § 429.510, and more specifically the adjustment described at proposed § 429.510(f). By collecting the MFP as part of the data submission requirement described at proposed § 429.615(b)(1), we may then consider the MFP as a component of the factor listed at section 1194(e)(1)(E) of the Act and the adjustment to the preliminary price described at proposed § 429.510(f)(2). We note that while the MFP is a publicly available price updated regularly by CMS, section 1194(e)(1) of the Act requires information related to the section 1194(e)(1) factors to be submitted by the Primary Manufacturer for consideration in offer development and during consideration of counteroffers, if applicable (as discussed in section II.F.2.a. of this proposed rule). Thus, collecting the MFP through the Drug Price Negotiation ICR (OMB 0938-1452) would allow us to consider the MFP within the context of the section 1194(e)(1) adjustment as discussed in proposed § 429.510(f)(2).
As an alternative to the proposal at § 429.620(d), we considered applying the policy set forth in the Negotiation Program Guidance. In this option we would not include the MFP as a component of the factor listed at section 1194(e)(1)(E) of the Act and therefore the MFP would not be considered within the section 1194(e)(1) adjustment described at § 429.510(f). With this alternative, we would also not collect the MFP through the Drug Price Negotiation ICR as the data would not be required for offer development or counteroffer consideration. We did not propose this option as we believe that the MFP is an important data point to consider within the context of renegotiation, as discussed previously.
As another alternative, we considered applying a third and separate adjustment based on the prior agreed upon MFP after the adjustment based on the section 1194(e)(1) factors is applied to the preliminary price as described at proposed § 429.510(f)(2). We believe this alternative would also be consistent with the methodology and process for the negotiation process to the extent practicable, but which would incorporate additional adjustments for the renegotiation context to afford distinct weight to the prior negotiation. In this approach, the MFP would be collected through the data submission requirement described at proposed § 429.615(b)(1) but considered separately from other information collected related to the section 1194(e)(1) factors. For example, for drugs selected for negotiation for initial price applicability year 2028, the Primary Manufacturer and CMS may have conducted up to three negotiation meetings following CMS' provision of the initial offer. During those meetings, the Primary Manufacturer may have, for example, presented additional evidence or otherwise raised a point not previously considered that contributes to our determination to accept a counteroffer or provide a subsequent offer that is different than the initial offer. If we were to follow the initial offer development process as described in proposed § 429.510 without considering the information that may have been exchanged over the course of the negotiation (or prior renegotiation, if applicable), then there may be circumstances where the initial offer is similar to the initial offer from the previous negotiation or renegotiation, and does not account for the additional discussions and information that contributed to the agreed upon MFP. By adjusting the amount determined after the section 1194(e)(1) adjustment described at proposed § 429.510(f)(2) by the MFP, we would be able to consider such negotiation meeting discussions and provide an initial offer that is informed by the prior negotiation or renegotiation process. We acknowledge that this approach would consider the MFP separately from the other section 1194(e)(1) factors. However, we believe this alternative approach would be consistent with the negotiation process to the extent practicable per sections 1194(f)(4)(B) and 1194(b)(1) of the Act as the initial offer development process would otherwise remain the same except for the single MFP adjustment.
d. Engagement With Primary Manufacturers and Interested Parties
We propose at § 429.620(e) through (l) that the renegotiation process would conform to the same procedures, structure, and timeline set forth for the negotiation process to the extent practicable. We note that we would apply the ceiling calculated for renegotiation per proposed § 429.615(b) when adopting these processes where applicable. We may incorporate drugs selected for renegotiation into the public engagement events for drugs selected for
( printed page 36309)
negotiation or host separate events specifically for drugs selected for renegotiation, as described in § 429.620(e).
If the Primary Manufacturer is delayed in meeting one or more deadlines in proposed §§ 429.525 through 429.535, such as submitting the renegotiation written counteroffer, we would continue to engage in the renegotiation process and would complete the established process as described in section II.C.6. of this proposed rule and as proposed subpart F. In the circumstance that CMS and the Primary Manufacturer do not agree to an MFP by the deadline set forth in proposed § 429.535, we refer to the discussion and proposal in subpart K, as described in section II.C.7. of this proposed rule. Additionally, if the failure to meet a deadline results in the failure of CMS and the Primary Manufacturer to reach an MFP following renegotiation, this may result in certain sales of the selected drug being subject to a potential excise tax (see 26 U.S.C. 5000D(b)(3)).
e. Publication of the MFP
For a selected drug for which CMS and a Primary Manufacturer agree upon an MFP through the renegotiation process, we propose at § 429.620(k) to publish and update the renegotiated MFP and related information as proposed in § 429.705 and as described in section II.H.2. of this proposed rule.
We do not intend to include redacted information from any voluntary information submitted by a Primary Manufacturer in response to the Drug Selection ICR in the MFP explanation if the selected drug of the Primary Manufacturer is selected for renegotiation and there is an agreement upon a renegotiated MFP. If the selected drug is then selected for renegotiation and the Primary Manufacturer submits the same information the Primary Manufacturer provided in response to the Drug Selection ICR also provided in response to the Drug Price Negotiation ICR, as described in section II.G.4. of this proposed rule and in proposed § 429.615, we may redact and include information provided in response to the Drug Price Negotiation ICR in the MFP explanation of a renegotiated MFP as proposed in section II.H.2. of this proposed rule and § 429.705(b) and in accordance with section II.H.2. of this proposed rule and proposed § 429.705(c) and the confidentiality policy described in section II.D.1. of this proposed rule and in proposed § 429.300.
H. Implementation of the MFP (§§ 429.700 Through 429.710)
1. Application of the MFP Across Dosage Forms and Strengths (§ 429.700)
Section 1196(a)(2) of the Act states an administrative duty in which we must establish procedures to compute and apply the MFP across different strengths and dosage forms of a selected drug and not based on the specific formulation or package size or package type of such drug. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 60.5 of the Negotiation Program Guidance with respect to initial price applicability year 2028. We propose at § 429.700 to codify the administrative duty requirement and that once the MFP has been agreed upon for a selected drug, we would compute and apply the MFP across different dosage forms and strengths of a selected drug. We propose at § 429.700 the methodology that we would use to apply the single agreed-upon MFP (which, as proposed at § 429.415 and section II.E.4. of this proposed rule, would be an average price per 30-day equivalent supply for the selected drug across all formulations of the selected drug) across NDC-9s and HCPCS codes, as applicable, and calculate an MFP-per-billing unit price for each billing unit and payment code associated with the selected drug, as applicable, as contemplated under section 1196(a)(2) of the Act and described at § 429.700(b)(4). CMS proposes to use a methodology that scales the MFP per unit and the MFP per billing unit based on price differentials across different dosage forms and strengths of the selected drug to ensure that the MFP is made available to MFP-eligible individuals at the point of sale (and to dispensing entities and Part B providers), we would publish the MFP per NCPDP unit (for example, tablet) for each NDC-9 and per billing unit for each HCPCS code, as applicable, associated with the selected drug based on the list of NDCs and HCPCS codes determined as described at § 429.100(c). We advise supply chain entities to use the NDC-9 MFP per unit price when effectuating the MFP for a selected drug with a formulation covered under Part D to ensure accuracy (for example, in the event of partial package dispense).
As proposed at § 429.700(b), we describe the proposed procedures to compute and apply the MFP across dosage forms and strengths of the selected drug using the WAC of the selected drug as reported by Primary Manufacturer in the CMS HPMS (as described in proposed § 429.20). This proposed process would apply the MFP to any NDCs of the selected drug, to include NDCs of a self-administered drug, assigned to the Primary Manufacturer and/or Secondary Manufacturer(s) where such NDCs do not represent sample packages and where the Primary Manufacturer reported a non-zero WAC for at least one calendar quarter of the calendar year 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation. (The proposed process for applying the MFP to NDCs of the selected drug with insufficient data is set forth in proposed § 429.700(c) as described below.) For NDCs of selected drugs covered under Part D, we would use PDE records from the calendar year that is 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation where the PDE record is associated with a prescription filled between January 1st of that year and December 31st of that year, and meets the inclusion criteria proposed at § 429.120(a)(2) through (a)(5) (as discussed in section II.B.5.a. of this proposed rule). With respect to NDCs of selected drugs payable under Part B, we would use OM Part B claims data and/or MA encounter data for Part B items and services from the calendar year 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation where the claim and/or record is associated with a service date between January 1st of that year and December 31st of that year, and meets the inclusion criteria proposed in proposed at § 429.120(b)(1)(ii) through (b)(1)(vi) (as discussed in section II.B.5.b. of this proposed rule). We propose to use the calendar year 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation for purposes of the calculations set forth in § 429.700(b) because it will be the most recent period of data available.
We propose at § 429.700(b)(1)(i), for each NDC-11 and calendar quarter, to calculate the quarterly WAC per unit by dividing the WAC quarterly units by the total WAC annual units (from the manufacturer-submitted data) and multiply this quotient by the quarterly WAC per unit. At § 429.700(b)(1), we propose to use the WAC unit cost for the period beginning on January 1 and ending December 31 for the year that begins 3 years prior to the initial price applicability year with respect to which the selected drug was selected for
( printed page 36310)
negotiation. We propose at § 429.700(b)(1)(ii), for each NDC-11, to sum the results determined under § 429.700(b)(1)(i) and to calculate the annual WAC per-unit cost for each of the NDC-11s for the selected drug (including NDC-11s payable under Part B, which are assigned to HCPCS codes in NDC-HCPCS code crosswalk files published by CMS, as well as NDC-11s covered under Part D) from the manufacturer-submitted quarterly WAC per unit and unit volume data to account for potential variation in unit volume across quarters.
We propose at § 429.700(b)(2) to convert the annual WAC per unit for each NDC-11 into an amount for a 30-day equivalent supply (using the methodology described § 423.104(d)(2)(iv)(A)(2) for Part D and at § 429.415(a)(2)(v) for Part B), so that the WAC will be comparable to the negotiated single MFP. We propose in § 429.700(b)(2)(i) to determine total units for the purpose of this calculation as the sum of the total quantity dispensed for NDC-11s present only in PDE data and the NDC-11 NCPDP unit equivalent conversion of the total Part B billing units administered for NDC-11s associated with HCPCS codes present in Part B data.
We propose at § 429.700(b)(3)(i) through (b)(3)(v), to then aggregate the WAC per 30-day equivalent supply for each NDC-11 into a WAC per 30-day supply for each NDC-9 of the selected drug. The WAC per 30-day equivalent supply for each NDC-9 would then be used to calculate a WAC price ratio for each NDC-9 of the selected drug as proposed in § 429.700(b)(3)(vi). The WAC price ratio derived from the WAC per 30-day equivalent supply for each NDC-9 would then be multiplied by the single MFP for the selected drug to calculate the MFP for a 30-day equivalent supply of each NDC-9 of the selected drug as proposed in § 429.700(b)(4)(A).
Lastly, we propose at § 429.700(b)(4)(i)(B), we would convert from an MFP for a 30-day equivalent supply to an MFP per NCPDP unit based on the average number of NCPDP units in a 30-day equivalent supply to determine the per NCPDP unit MFP for an NDC-9. We propose at § 429.700(b)(4)(ii), that for selected drugs payable under Part B, we would further convert the MFP per NCPDP unit of each NDC-9 into an MFP per billing unit by converting the per-NCPDP unit amount into an amount per billing unit as necessary to account for any differences in the NDC-9 unit versus the billing unit. This conversion would require us to convert the NDC-9 MFP per NCPDP unit to an NDC-11 MFP per package (by multiplying the NDC-9 MFP per NCPDP unit by the NDC-11 NCPDP package quantity), and then divide this product by the HCPCS billing units per NDC-11 package to arrive at an NDC-11 MFP per billing unit. We would then take an average of the NDC-11 MFP per billing unit across all NDC-11s associated with a HCPCS code for the selected drug, weighted by the ASP units reported by manufacturers for each NDC-11 under section 1847A of the Act.
We would include the MFP per billing unit, calculated at § 429.700(b)(4)(ii)(E), and the MFP per NCPDP unit price for each NDC-9 of the selected drug, calculated at § 429.700(b)(4)(i)(B) in the publication of MFPs as proposed in § 429.705. We recognize there may be other ways to apply the MFP to dosage forms and strengths and intend to monitor this policy. We propose to codify the approach taken in policies for implementation as described in section 60.5 of Negotiation Program Guidance to list in the public pricing file only the NDC-9 MFP-per-unit price (rounded to six decimals) rather than also including the NDC-11 MFP-per-package price. The NDC-11 MFP-per-package price was removed due to the potential for confusion arising from differences between the NDC-11 MFP-per-package price as published by CMS (calculated without rounding any values in interim steps) and the NDC-11 MFP-per-package price that would result if another entity were to take the product of the published NDC-9 MFP-per-unit price (rounded to six decimals) and the package size. The application of the MFP is not inclusive of NDCs of the selected drug that are not manufactured, marketed, controlled, or sold by the Primary Manufacturer or a Secondary Manufacturer, or NDC-11s to which the MFP would not apply, such as sample packages, inner packages, and discontinued products.
Consistent with policies for implementation as described in section 60.5.1 of Negotiation Program Guidance, we propose at § 429.700(c), the process by which we would apply the MFP to NDCs associated with new NDAs/BLAs, NDCs, or HCPCS codes, including those added during the negotiation period or after agreeing upon an MFP, and to NDCs and HCPCS codes with insufficient PDE, Part B data, or WAC data in calendar year 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation to apply the MFP across those dosage forms and strengths. We propose at § 429.700(c)(1), the process for how we would apply the MFP to NDCs associated with new NDAs or BLAs and we propose at § 429.700(c)(2), the process for how we would apply the MFP to NDCs associated with HCPCS codes of new NDAs or BLAs. We propose at § 429.700(c)(3), the process for how we would apply the MFP to NDCs and HCPCS codes with insufficient data, which would be considered if there was no data available to calculate a sum of the plan-specific enrollment weighted amount in the applicable calendar year, no payment amount under section 1847A(b)(4) of the Act, and no average non-FAMP data (when it has not yet been a full year following the market entry for such drug) are determined to have insufficient data. As proposed at § 429.700(c)(4)(i), we would determine whether there is an existing NDC that is comparable to the new NDC and has sufficient data for the MFP application calculations proposed § 429.700(b). We would base our review of a comparable NDC using the FDA-approved label of the selected drug and other relevant sources. We would evaluate whether an existing NDC is comparable to a new NDC if it shares the same NDC-9, has a different NDC-9 but has the same dosage form and strength, has a different NDC-9 but has a similar dosage form and strength, or has the same active moiety/active ingredient. We propose at § 429.700(c)(3)(i)(A) and (B), respectively, the process that we would use if an existing, comparable NDC exists and the process if a comparable NDC does not exist.
We propose at § 429.700(c)(3)(ii), the process for which we would adjust the MFP application. Specifically for the NDCs and HCPCS codes described in § 429.700(c)(3)(ii), we would monitor total quantity dispensed or administered and 30-day equivalent supply from PDE data and Part B data over time, beginning when these NDCs and/or HCPCS codes are first added to our computation of how we will apply a single MFP across dosage forms and strengths of the selected drug. We would update the total quantity dispensed and 30-day equivalent supply values and recompute the application of the single MFP across dosage forms and strengths for these NDCs and/or HCPCS codes (but only for such NDCs and/or HCPCS codes, not for all NDCs and/or HCPCS codes of the selected drug) based on which of the following situations occurs first: (1) a year has elapsed since the NDCs first appeared in PDE records or the HCPCS codes associated with the NDCs appeared in Part B data; or (2) the NDC or HCPCS
( printed page 36311)
code has accrued the same number of units dispensed/administered as the NDC-11 that had the fewest units dispensed/administered at the time that the WAC ratios were originally calculated. We included a third condition in section 60.5.1 of the Negotiation Program Guidance: CMS determines the variation in average total quantity dispensed and 30-day equivalent supply is stable over time. We propose to remove this condition due to its subjectivity and lack of predictability compared to the other two conditions and codify proposed § 429.700(c). We propose at § 429.700(c)(3)(iii), the process we would apply if a new NDC is assigned to a HCPCS code for which we have already calculated an MFP per billing unit.
We propose at § 429.700(d), that after we recompute the application of the single MFP across dosage forms and strengths for the new NDC-11 as proposed in § 429.700(c)(4)(i)(B)(1), to provide Primary Manufacturers with the calculations described at proposed § 429.700(b) and (c) and if the Primary Manufacturer believes in good faith that we made an error, a Suggestion of Error may be submitted, as set forth in § 429.445(c), in a form and manner as specified by CMS, as set forth in § 429.445(d), as described in section II.E.11. of this proposed rule. We solicit comments on our proposed approaches for the application of the MFP across dosage forms and strengths.
2. Publication of the MFP (§ 429.705)
Section 1195(a)(1) of the Act requires that with respect to an initial price applicability year and a selected drug with respect to such year, not later than November 30 of the year that is 2 years prior to such initial price applicability year, CMS shall publish the MFP for such drug negotiated with the manufacturer of such drug. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for example, section 60.6 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, and consistent with section 60.6 of the Negotiation Program Guidance, we propose at § 429.705(a) that we would publish by November 30 of the year that is 2 years prior to the initial price applicability year, the MFP for each selected drug for which CMS and the Primary Manufacturer have reached an agreement on an MFP. For example, we would publish the MFP for each selected drug for which CMS and the Primary Manufacturer have reached an agreement on an MFP no earlier than November 1, 2027 and no later than November 30, 2027 for initial price applicability year 2029. Related to this requirement, we propose at § 429.705(a)(1) to publish the following on the CMS website: the selected drug; the initial price applicability year; and the MFP file. The MFP file [64]
would contain the single MFP for a 30-day equivalent supply of the selected drug, NDC-9 MFP-per-unit price and HCPCS code dosage price and would be updated annually to show the inflation-adjusted MFP for the selected drug. We would also update the file as needed if any NDC-9s or HCPCS codes are added or removed for the selected drug, or if the NDC-9 MFP per-unit price or HCPCS code dosage price is updated as a result of additional data.
Further, as proposed at § 429.705(a)(1)(iv), we would publish on the CMS website whether an MFP between a Primary Manufacturer and CMS is not agreed upon. As proposed at § 429.705(a)(1)(v), we would also publish on the CMS website whether a drug is no longer a selected drug and the reason for that change. In accordance with section 1192(c) of the Act, a selected drug with an agreed-upon MFP would cease to be a selected drug and no longer be subject to an MFP in accordance with the timeline described in section II.B.6.d. of this proposed rule and in proposed subpart B if we determine that a generic drug or a biosimilar for the selected drug is approved or licensed by the FDA and—as proposed under subpart B—is subject to Bona Fide Marketing. We further recognize that, in accordance with section 1194(f) of the Act, the MFP for a selected drug may also change due to renegotiation beginning in initial price applicability year 2028 (in the case of a renegotiation-eligible drug selected by the Secretary under section 1194(f)(3) of the Act), as described in section II.G. of this proposed rule and in proposed subpart G.
Section 1195(b)(1)(A) of the Act requires that, for a selected drug for each year subsequent to the first initial price applicability year of the price applicability period with respect to such drug, with respect to which an agreement for such drug is in effect under section 1193 of the Act, not later than November 30 of the year that is 2 years prior to such subsequent year, CMS shall publish the MFP applicable to such drug and year, which shall be the amount equal to the MFP published for such drug for the previous year, increased by the annual percentage increase in the consumer price index for all urban consumers (all items; United States city average) for the 12-month period ending with the July immediately preceding such November 30. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for example, section 60.6 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, and consistent with section 60.6 of the Negotiation Program Guidance, we are proposing in § 429.705(a)(2) that for each selected drug, for each year subsequent to the first initial price applicability year of the price applicability period (unless renegotiation occurs as set forth in section II.G. of this proposed rule and under proposed subpart G), we would publish an updated MFP no later than November 30 of the year that is 2 years prior to such subsequent year. We propose at § 429.705(a)(2)(i) that the updated MFP for each selected drug would be equal to the MFP that was published for such drug for the previous year, increased by the annual percentage increase in the CPI-U for the 12-month period ending with the July immediately preceding such November 30. For example, no later than November 30, 2028, we would publish on the CMS website updated amounts for any MFPs for initial price applicability year 2029 selected drugs for which a manufacturer agreement is in effect. Those updated MFPs would take effect in 2030 and would be equal to the initial price applicability year 2029 MFP for the selected drug increased by the percent increase in CPI-U from July 2027 to July 2028.
Section 1195(b)(2) of the Act requires that, in the case of a selected drug with respect to an initial price applicability year for which the MFP is determined after the date of publication under section 1195(a) of the Act, CMS shall publish such MFP by not later than 30 days after the date such MFP is so determined. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for
( printed page 36312)
example, section 60.6 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, and consistent with section 60.6 of the Negotiation Program Guidance, we propose in § 429.705(a)(3) that in the case of a selected drug with respect to an initial price applicability year for which the MFP is determined after the MFPs are published for other selected drugs, such as due to the circumstances described in proposed § 429.710, we would publish the MFP no later than 30 days after the date such MFP is so determined.
Section 1195(a)(2) of the Act requires that with respect to an initial price applicability year and a selected drug with respect to such year, not later than March 1 of the year prior to such initial price applicability year, the Secretary shall publish, subject to section 1193(c) of the Act, the explanation for the MFP with respect to the factors as applied under section 1194(e) of the Act for such drug. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for example, section 60.6 of the Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, and consistent with section 60.6 of the Negotiation Program Guidance, we propose in § 429.705(b) that we would publish explanations for the MFPs no later than March 1 of the year prior to the initial price applicability year. We propose in § 429.705(b)(1) to develop and publish the explanations for the MFPs for each selected drug, or drug selected for renegotiation, subject to the requirements for treatment of confidential and proprietary information in proposed § 429.300 and described in section II.D.1. of this proposed rule. The explanation for the MFP includes: the narrative explanation of the MFP; redacted information regarding the negotiation meetings, as applicable, including exchanges of offers and counteroffers, as applicable; and the redacted information submitted by a Primary Manufacturer in proposed § 429.505(b)(2) or § 429.615(b)(1) (as described in section II.F.2. or section II.G.4. of this proposed rule), as applicable, and the redacted information submitted by interested parties in proposed § 429.505(d)(3) or § 429.615(b)(3), as applicable.
Within the explanation of the MFP, we may also make public, high-level comments about the sections 1194(e)(1) and 1194(e)(2) data submitted to CMS that are determined to be proprietary, without sharing any PHI/PII or any proprietary information reported to CMS under section 1193(a)(4) of the Act for purposes of the negotiation. Similar to the approach taken for publication of the public MFP explanations for initial price applicability year 2026 and 2027, for each drug, we would make available on the CMS website redacted versions of section 1194(e)(2) data that are determined to be nonproprietary and will not disclose any PHI, PII, or information that is protected from disclosure under other applicable law.
If an agreement for an MFP is reached for a selected drug and CMS makes a determination before the end of the negotiation period (as set forth in proposed § 429.535) that an approved generic drug or licensed biosimilar for the selected drug is subject to Bona Fide Marketing (consistent with proposed § 429.130(a)), we would neither publish an MFP nor publish an MFP explanation because the selected drug ceases to be subject to the negotiation, pursuant to section 1192(c)(2) of the Act and as described in proposed § 429.135(b)(1) (see further discussion in section II.B.6.d. of this proposed rule).
If an agreement for an MFP is not reached for a selected drug, we would neither publish an MFP nor publish an MFP explanation, as proposed at § 429.705(b)(2). Instead, we would indicate on the CMS website that an MFP has not been agreed upon between the Primary Manufacturer and CMS for the selected drug.
3. Establishment of MFPs After the Negotiation Deadline (§ 429.710)
Section 1194(b)(2) of the Act contemplates that agreement upon an MFP must be reached prior to November 1 following the selected drug publication date, with respect to the initial price applicability year, to avoid potential imposition of an excise tax. If negotiations have not ended by this date, the Primary Manufacturer may be subject to an excise tax under 26 U.S.C. 5000D. As a general matter, if the Primary Manufacturer is delayed in meeting one or more deadlines related to the negotiation process, we would continue to engage in the negotiation process described in section II.F. of this proposed rule and under proposed subpart F.
Certain actions or delays by the Primary Manufacturer may delay the process such that the MFP may be agreed to after the end of the negotiation period as described in proposed § 429.535(b). Section 1194(b)(1) of the Act requires that the Secretary shall develop and use a consistent methodology and process, in accordance with section 1194(b)(2) of the Act, for negotiations under section 1194(a) of the Act that aims to achieve the lowest MFP for each selected drug. With respect to initial price applicability years 2026 through 2028, we implemented this requirement through guidance, including, for example, Negotiation Program Guidance with respect to initial price applicability year 2028.
With respect to initial price applicability year 2029 and subsequent years, we propose in § 429.710 that, in the event of a delay by the Primary Manufacturer such that the MFP may be agreed to after the end of the negotiation period described in the proposed § 429.535(b), we would follow timelines consistent with the negotiation process established in section II.F. of this proposed rule and under proposed subpart F and take the time to complete the established process so described as appropriate for the selected drug. Certain actions by the Primary Manufacturer may delay the negotiation process to such an extent that a selected drug has a change in status that is material to CMS' statutory obligations under the negotiation process. If this occurs, as proposed at § 429.710(a), when CMS initiates or resumes the negotiation process, we would apply the consistent methodology and process with respect to the selected drug based on its status at the time the negotiation process occurs, including with respect to renegotiation, as applicable, as described in subpart G.
If the manufacturer and CMS complete each step of the negotiation process as described in section II.F. of this proposed rule and under subpart F, including CMS' issuance of a final offer in accordance with § 429.535(a), and then, after the statutory end of the negotiation period, the Primary Manufacturer of a selected drug wishes to agree to an MFP, the Primary Manufacturer must notify CMS in writing that it would like to accept the final offer from CMS, as proposed at § 429.710(b).
In accordance with section 1195(b)(2) of the Act and as proposed at § 429.705(a)(3), in the case of a selected drug with respect to an initial price applicability year for which the MFP is determined after the MFPs are published for other selected drugs, we would publish the MFP no later than 30 days after the date such MFP is so determined. For such a drug, in accordance with §§ 429.705(b) and 429.710, we would follow timelines consistent with the established process for publishing the explanation of the
( printed page 36313)
MFP and would not expedite our timeline due to late action from the Primary Manufacturer.
I. Manufacturer Compliance and Oversight (§ 429.900)
Section 1196(b) of the Act requires that CMS monitor compliance by a Primary Manufacturer with the terms of the Negotiation Program Agreement and establish a mechanism through which violations of such terms shall be reported. With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 90.1 of the Negotiation Program Guidance with respect to initial price applicability year 2028. With respect to initial price applicability year 2029 and subsequent years, we are proposing to codify our policies for effectuating the compliance monitoring required by section 1196(b) of the Act, consistent with those policies described in section 90.1 of the Negotiation Program Guidance, in proposed subpart J. Section 429.900 codifies sections 90 through 90.1 of the Negotiation Program Guidance.
1. Monitoring Manufacturer Compliance (§ 429.900)
Proposed § 429.900 establishes CMS' approach to monitoring and assessment of Primary Manufacturer compliance with the terms of the Negotiation Program Agreement as described in § 429.200, the Primary Manufacturer's obligation to cooperate with CMS's compliance monitoring activities, and actions CMS may take to address Primary Manufacturer noncompliance.
In accordance with section 1193(a)(5) of the Act, section 429.900(a) proposes that CMS may monitor and assess Primary Manufacturer compliance with the Negotiation Program Agreement, including compliance with all applicable requirements and conditions set forth in sections 1191 through 1198 of the Act and all applicable guidance and regulations, including part 429, implementing those provisions and any changes to the Act that affect the Negotiation Program. We will closely monitor the Primary Manufacturer's compliance with the terms of the Negotiation Program Agreement, including the requirements for Primary Manufacturers of selected drugs as described in part 429. Following the publication of selected drugs for negotiation and renegotiation for each initial price applicability year, as described in § 429.100, we may provide information about the negotiation or renegotiation process, as applicable, to the Primary Manufacturer of each selected drug. We anticipate this information would include operational and statutory timelines, procedural requirements, systems instructions, IRA resources, and contact information. During the negotiation and renegotiation periods, we would track and monitor progress during all steps of the process. CMS compliance monitoring will continue beyond the negotiation period and extend to all aspects of Primary Manufacturer participation in the Negotiation Program. For example, during the negotiation period and after it has closed, we may require additional information from the Primary Manufacturer to administer or monitor compliance with the Negotiation Program in accordance with section 1193(a)(5) of the Act. This may include requiring recurring reporting (for example, providing evidence that the MFP is being made available), or making specific ad hoc requests to the Primary Manufacturer for information related to targeted monitoring, auditing, or investigation efforts. Methods CMS may use to monitor and assess compliance, during the negotiation and renegotiation periods or otherwise, include but are not limited to: evaluation of complaints made by individuals and entities to CMS; CMS' engagement in direct communications with the Primary Manufacturer; and CMS audits and comprehensive reviews.
As described in proposed § 429.900(b), Primary Manufacturers must cooperate with CMS compliance monitoring activities. This includes providing complete, accurate, and relevant responses to CMS requests for clarifications, corrections, and additional information and complying fully with requests for corrective action during the compliance monitoring process and when noncompliance is identified. For example, we propose that Primary Manufacturers must submit complete, accurate, and relevant responses, in the form and manner and on the timeline specified by CMS, in response to written requests CMS provides to the Primary Manufacturer in the course of our compliance monitoring activities when we deem appropriate. We are committed to providing Primary Manufacturers with reasonable timeframes to accommodate these information requests; Primary Manufacturers have the burden of establishing why an extension of any deadline to submit information should be considered by CMS. If a Primary Manufacturer articulates a reasonable basis for seeking an extension for a reasonable duration of time, and the request is submitted at a reasonable point in time before the deadline (for example, 3 calendar days prior to the initial deadline), then CMS may grant an extension of a limited duration to the extent consistent with statutory timelines and other operational considerations.
Likewise, we propose that a Primary Manufacturer must comply fully with corrective action requests that we provide to the Primary Manufacturer when we deem it appropriate; for example, including but not limited to, in the event that a Primary Manufacturer fails to submit data as described in proposed §§ 429.100 and 429.505 of this proposed rule. We recognize the substantial role that manufacturer-submitted information will play in the negotiation and renegotiation processes and the need for complete and accurate information. Should CMS determine that a submission is incomplete or contains inaccurate information, CMS may provide a written request to the Primary Manufacturer to clarify the submission, correct the inaccuracy, or provide the necessary information, with a deadline by which the Primary Manufacturer must respond. The written corrective action request would outline the needed action and establish a deadline for the Primary Manufacturer to correct the submission and/or provide additional information to validate the accuracy and completeness of the original submission. We intend to be available to engage with the Primary Manufacturer about the specifics of a corrective action request and to answer questions and provide clarification.
The information required in this proposed part is information required by CMS to administer and monitor the Negotiation Program in accordance with section 1193(a)(5) of the Act. As such, as proposed at § 429.900(b), failure to provide complete and accurate information, initially or in response to CMS' initial questions or corrective action requests, may result in the Primary Manufacturer being subject to a civil monetary penalty as authorized under section 1197(c) of the Act and as described in proposed § 429.1005.
In proposed § 429.900(c), we would establish the actions CMS may take if we conclude that a Primary Manufacturer is noncompliant with one or more requirements of the Negotiation Program Agreement, including all applicable requirements and conditions set forth in sections 1191 through 1198 of the Act and all applicable guidance and regulations, including part 429, implementing those provisions and any changes to the Act that affect the Negotiation Program. Upon identifying a
( printed page 36314)
violation, CMS may take one or more of the following actions: (1) CMS may provide a written notice to the Primary Manufacturer of the violation; (2) CMS may request the Primary Manufacturer to take specific corrective action to address the noncompliance; or (3) CMS may impose a civil monetary penalty on the Primary Manufacturer as set forth in subpart K. In instances in which CMS provides a written notice to the Primary Manufacturer of the violation and/or requests specific corrective action, the agency may offer the Primary Manufacturer the opportunity, by a specified deadline, to provide information regarding the circumstances of violation, evidence seeking to refute the finding of violation, proof of mitigation of noncompliance, and other factors for CMS' consideration. CMS proposes to consider such information if timely submitted when determining whether to pursue further enforcement action such as imposition of civil monetary penalties.
J. Civil Monetary Penalties (§§ 429.1005 Through 429.1020)
1. Civil Monetary Penalties
Section 1197 of the Act provides for the imposition of civil monetary penalties on manufacturers for: (1) failure to provide access to a price that is less than or equal to the maximum fair price for a drug; (2) failure to pay the rebate amount for a biological product inclusion of which on the selected drug list was delayed but has since undergone negotiation, as described in section 1192(f)(4) of the Act; (3) violation of certain terms of the Negotiation Program Agreement; and (4) the provision of false information as described in section 1197(d) of the Act.
With respect to initial price applicability years 2026 through 2028, we implemented these requirements through guidance, including, for example, section 100 of the Negotiation Program Guidance with respect to initial price applicability year 2028. With respect to initial price applicability year 2029 and subsequent years, with the exception of our authority pursuant to section 1197(a) of the Act as described later in this section, we are proposing to codify our policies for implementation of our CMP authorities as set forth in section 1197 of the Act, consistent with those policies described in section 100 of the Negotiation Program Guidance, in proposed subpart K.
In future rulemaking, we will codify our policies for implementation of section 1197(a) of the Act, under which manufacturers may be subject to a civil monetary penalty for failure to provide access to a price less than or equal to the MFP, for 2029 and subsequent years. Consistent with the statutory directive to use program instruction or other forms of program guidance to implement sections 1191 through 1198 of the Act for 2026, 2027, and 2028, a Primary Manufacturer's compliance obligations with respect to its obligation to provide access to a price less than or equal to the MFP for 2026, 2027, and 2028 are established in the applicable program guidance, and we are reserving codifying the civil monetary penalty associated with violation of this obligation, including the process, calculation, and substantive compliance obligations, for 2029 and subsequent years, for future rulemaking.
In proposed § 429.1005, we address potential imposition of civil monetary penalties for a Primary Manufacturer's violation of a requirement determined by CMS to be necessary for the purposes of administering and monitoring compliance with the Negotiation Program, including without limitation, the requirement to submit information pursuant to section 1193(a)(4) of the Act, in accordance with section 1197(c) of the Act. In proposed § 429.1010, we address potential imposition of civil monetary penalties for provision of false information for use in applying the eligibility rules at sections 1192(d)(2)(B) and (f)(1)(C) of the Act, in accordance with section 1197(d) of the Act. In proposed § 429.1015, we address potential imposition of civil monetary penalties for failure to pay a rebate as required by section 1192(f)(4) of the Act, in accordance with section 1197(b) of the Act. In proposed § 429.1020, we address details about the process for imposition of civil monetary penalties. Separate from CMS imposition of CMPs, per 26 U.S.C. 5000D, failure of a Primary Manufacturer to comply with certain Negotiation Program deadlines and other requirements of the Negotiation Program may result in potential excise tax liability.
Our primary goal is to successfully administer all aspects of the Negotiation Program; we intend to exercise the authority to impose civil monetary penalties in instances of noncompliance that substantively obstruct Negotiation Program processes. Such instances may include, but are not limited to, the examples shown in Table 4, such as failure to provide timely, complete, and accurate information that is necessary to execute the negotiation process or other administrative or monitoring functions of the Negotiation Program; repeated violations of the Negotiation Program Agreement or other Negotiation Program requirements determined by CMS to be necessary for the purposes of administering and monitoring compliance with the Negotiation Program; or egregious or knowing violations of Negotiation Program requirements. Note that these examples are not an exhaustive list of violations that could warrant civil monetary penalties. We reserve the authority to issue civil monetary penalties for other violations as required to effectively administer and monitor the Negotiation Program.
Table 4—Examples of Substantive Violations 65
Category
Example
Violations of the Negotiation Program Agreement
• Failure to submit data required under proposed §§ 429.100(d), 429.405(a), and 429.505(b)(2) including failure to engage in requested corrective action to mitigate such failures.
• Omission or inaccuracy of manufacturer-submitted information described in proposed §§ 429.100(d), 429.405(a) and 429.505(b)(2) (for example, non-FAMP data from the Primary Manufacturer, including non-FAMP data for a selected drug manufactured, marketed, controlled, or sold by any Secondary Manufacturer(s), required for ceiling calculation) or other violations that impede CMS efforts to administer or monitor the Negotiation Program (for example, failure to report new NDC-11s, failure to provide information requested during an audit) as well as failure to engage in requested corrective action to mitigate such omissions, inaccuracies or other violations.
• Submission of false information that interferes with the negotiation process (for example, submission of false data on unit costs of production or research and development costs).
( printed page 36315)
• Failure to provide information requested by CMS in accordance with CMS' oversight responsibilities under section 1196(b) of the Act and described in subpart J.
Other Violations
• Knowing provision of false information to CMS for use in applying the aggregation rule described at § 429.110(b)(1)(iv)(A).
• Knowing provision of false information to CMS for use in applying the test to determine if a selected drug or drug selected for renegotiation is eligible for the Temporary Floor for Small Biotech Drugs as described in proposed § 429.440(b)(2).
• Failure to pay a biosimilar delay rebate by the deadline established in proposed § 429.110(h).
Broadly, we propose to establish a structure for enforcement actions that: (1) is within CMS' statutory authority; (2) is not punitive in response to immaterial noncompliance, or other instances of noncompliance that are not substantive; (3) can be applied consistently across applicable instances of Primary Manufacturer noncompliance; and (4) facilitates the ability to successfully engage in all components of the negotiation process within the established timeframes.
2. Violations of the Negotiation Program Agreement (§ 429.1005)
In accordance with section 1197(c) of the Act, any Primary Manufacturer of a selected drug that has entered into an Negotiation Program Agreement with CMS as set forth in proposed § 429.200 that fails to comply with requirements determined by CMS to be necessary for the purposes of administering the Negotiation Program and monitoring compliance with the Negotiation Program, including failure to provide information required under section 1193(a)(4) of the Act, including information required to be submitted under proposed §§ 429.405(a) and 429.505(b)(2), may be subject to statutorily-specified civil monetary penalties for each day of such violation. The statutorily specified amount of $1,000,000 a day is updated yearly per the Federal Civil Penalties Inflation Adjustments Improvements Act of 2015. In applying civil monetary penalties for Primary Manufacturer violations of the Negotiation Program Agreement, we intend to use discretion such that civil monetary penalties are reserved for instances of substantive noncompliance.
Effective oversight of the Negotiation Program requires that CMS receive complete and accurate data from Primary Manufacturers. We view the authority to impose civil monetary penalties as established in section 1197(c) of the Act as a tool to ensure that a Primary Manufacturer is participating in the Negotiation Program consistent with its signed Negotiation Program Agreement, including, but not limited to, providing complete and accurate information CMS deems necessary for administering and monitoring compliance with the Negotiation Program and promptly responding to any communications from CMS regarding the Negotiation Program, including requests for clarification, correction, additional information, or specific corrective actions.
In this proposed rule, we propose to consolidate and clarify our approach to monitoring and enforcement in the context of data reporting. Specifically, we are not proposing to codify the policies previously stated in section 40.2.3 of the Negotiation Program Guidance but rather are pursuing a consolidated approach to monitoring and enforcement that is consistent with the policies regarding data reporting obligations previously stated in sections 90 and 100 of the Negotiation Program Guidance. For example, we may impose a civil monetary penalty for violation of a requirement of the Negotiation Program Agreement if a Primary Manufacturer fails to provide data required under the Drug Price Negotiation ICR Forms, such as information on non-FAMP for each applicable quarter, as described in proposed § 429.405 of this proposed rule, for each NDC-11 of the selected drug for the applicable period, by the deadline established at proposed § 429.505(b)(1). In this scenario, upon identifying the missing data, we may take a variety of actions. For one, in accordance with proposed § 429.900(c), we may send the Primary Manufacturer a request for corrective action to address the noncompliance, such as a request that the Primary Manufacturer provide a complete and accurate production of the outstanding non-FAMP data by a specified date. If the Primary Manufacturer complies with the request for corrective action by timely providing complete and accurate non-FAMP data, we may exercise discretion and determine not to impose a civil monetary penalty for the Primary Manufacturer's initial failure to provide complete and accurate non-FAMP data by the specified deadline. Or, depending on the circumstances, in accordance with enforcement priorities, we may determine that, even if a Primary Manufacturer complies with a request for corrective action, it remains appropriate to issue a violation notice and impose a CMP for the initial violation. Alternatively, in accordance with proposed § 429.900(c), we may not issue a request for corrective action and instead send a notice of violation to the Primary Manufacturer and impose a CMP in accordance with subpart K. Finally, if we send the Primary Manufacturer a request for corrective action and the Primary Manufacturer does not comply with such request, in accordance with proposed § 429.900(c), we may issue a violation notice and impose a CMP for the failure to comply with CMS' corrective action request, which would be separate from any violation notice and CMP imposition addressing the violation that arose from the Primary Manufacturer's initial failure to provide complete and accurate non-FAMP data by the specified deadline.
In a case where a civil monetary penalty is pursued, we intend to send a written civil monetary penalty notification, as described in proposed § 429.1005(d), that reflects the start date of the penalty accrual, the end date of the penalty accrual, and the total amount of the penalty assessed, as described in proposed § 429.1020(a). As described at proposed § 429.1005(c), the state date of the penalty accrual is the first day of the violation as set forth in proposed § 429.1005(a). In the earlier example, the start date of the penalty accrual would be the day after the applicable submission deadline. As described at proposed § 429.1005(c), the civil monetary penalty shall accrue until the Primary Manufacturer has provided the necessary information or otherwise taken any corrective action determined by CMS to be necessary to address the violation, including, as applicable, providing documentation to evidence that the Primary Manufacturer has provided all past due information, or
( printed page 36316)
the Negotiation Program Agreement is terminated. In the earlier example, in the event the Primary Manufacturer never provides the required information, the daily civil monetary penalty would continue to accrue until the Negotiation Program Agreement was terminated as described in proposed § 429.205. We plan to adopt the same approach to enforcement in a circumstance where a Primary Manufacturer failed to provide data for a drug selected for renegotiation, with a violation accruing each day after the deadline established for provision of the data in proposed § 429.615 until the Primary Manufacturer provided the data. In the event the Primary Manufacturer never provides the required data, the civil monetary penalty will continue to accrue until the Negotiation Program Agreement is terminated as described in proposed § 429.205. The imposition of a civil monetary penalty related to failure to provide required data for the renegotiation process does not negate the Primary Manufacturer's ongoing obligation to provide access to the previously negotiated MFP. Primary Manufacturers continue to have the obligation to make the previously negotiated MFP available.
Another example of when CMS may impose a civil monetary penalty for violation of the Negotiation Program Agreement is if the Primary Manufacturer submits information that is required under the Negotiation Program Agreement and CMS determines the information is false. In this example, the Primary Manufacturer would be determined to be noncompliant with the requirement to submit information and may be subject to a civil monetary penalty. In instances of a Primary Manufacturer submitting false information that is required under the Negotiation Program Agreement, in accordance with proposed § 429.1005, a civil monetary penalty would begin to accrue on the day after the established deadline for submission of information under the Negotiation Program Agreement and would continue to accrue until the Primary Manufacturer provides a complete and accurate submission of the required information to CMS or the Negotiation Program Agreement is terminated as described in § 429.205. The start and end date of civil monetary penalty accrual as well as the total amount accrued will be noted on the civil monetary penalty notification sent by CMS, following the process established in proposed § 429.1020.
3. Provision of False Information Related to the Biosimilar Delay and Temporary Floor for Small Biotech Drugs (§ 429.1010)
In accordance with section 1197(d) of the Act, we are proposing at § 429.1010 two circumstances where CMS may impose a CMP due to provision of false information: (1) CMS may impose a statutorily specified civil monetary penalty on a Biosimilar Manufacturer for each item of false information the Biosimilar Manufacturer knowingly provides to CMS for use in applying the aggregation rule described at proposed § 429.110(b)(1)(iv)(A); and (2) CMS may impose a statutorily specified civil monetary penalty on a Primary Manufacturer for each item of false information the Primary Manufacturer knowingly provides to CMS for use in applying the test to determine if a selected drug or drug selected for renegotiation is eligible for the Temporary Floor for Small Biotech Drugs described at proposed § 429.440(b)(2). Section 1197(d) of the Act provides for imposition of a civil monetary penalty in the amount of $100,000,000 per item of false information. This amount will be updated yearly per the Federal Civil Penalties Inflation Adjustment Improvements Act of 2015.
CMS adopts a standard for “knowingly” that has the meaning set forth in 42 CFR 1003.110. Knowingly means that a manufacturer: (1) has actual knowledge of the information; (2) acts in deliberate ignorance of the truth or falsity of the information; or (3) acts in reckless disregard of the truth or falsity of the information. No proof of specific intent to defraud is required. Upon identifying instances of knowing submission of false information under this provision, CMS intends to provide the Biosimilar Manufacturer or Primary Manufacturer, as applicable, with a civil monetary penalty notification following the process established in proposed § 429.1020.
4. Failure To Pay a Biosimilar Delay Rebate (§ 429.1015)
In accordance with section 1197(b) of the Act, where a Reference Manufacturer fails to comply with the rebate requirements under section 1192(f)(4) of the Act as described at proposed § 429.110(i) and section II.B.3. of this proposed rule, CMS is proposing at § 429.1015 that the Reference Manufacturer may be subject to a civil monetary penalty equal to 10 times the amount of the rebate the Reference Manufacturer failed to pay. When CMS makes a determination to assess a civil monetary penalty under section 1197(b) of the Act, CMS intends to follow the procedures established in proposed § 429.1020.
5. Notice and Appeal Procedures (§ 429.1020)
Under section 1197(e) of the Act, civil monetary penalties imposed in accordance with sections 1197(a) through (d) of the Act are subject to the provisions of section 1128A of the Act (other than subsections (a) and (b)). Accordingly, when CMS makes a determination to assess a civil monetary penalty, we intend to provide a written civil monetary penalty notification that the manufacturer has engaged in one or more violations as described at proposed §§ 429.1005 through 429.1015 and is subject to a civil monetary penalty. We are proposing at § 429.1020(a) that the civil monetary penalty notification would include the following:
A description of the basis for the determination.
The basis for the penalty,
The start date of the penalty (if applicable).
The end date of the penalty (if applicable).
The total amount of the penalty assessed.
The date the penalty is due.
The manufacturer's right to a hearing.
Information about where to file the request for a hearing.
In the case of violations associated with civil monetary penalties with daily accruals as described in proposed § 429.1005, we intend to send the civil monetary penalty notification after the accrual has ended to reflect both the start date, end date, and total amount of penalty assessed within the notice. We have considered an alternative policy in which civil monetary penalty notification letters would be issued during the period that the daily penalty amounts are still actively accruing. Such an approach would result in assessment of interim civil monetary penalties on Primary Manufacturers, each for the dollar amount that had accrued as of the date we issue each applicable interim CMP notice. We would issue such interim notices at set time intervals (for example, every 14 days) throughout the period in which the penalty continues to accrue as described in proposed § 429.1005. CMS is soliciting comment on this alternative option.
Per section 1128A of the Act, civil monetary penalties are due 60 calendar days after the receipt of the civil monetary penalty notification, unless the manufacturer chooses to initiate an
( printed page 36317)
appeal. At the conclusion of any appeal process initiated by the manufacturer, where there is still a civil monetary penalty amount owed, the civil monetary penalty is due within 60 calendar days of the appeal decision.
To operationalize the civil monetary penalty appeal process in the Negotiation Program, we propose adopting the existing procedures as codified in 42 CFR part 423 subpart T: Appeal Procedures for Civil Money Penalties (see 42 CFR 423.1000 through 423.1094) that currently apply to Part D sponsors and to manufacturers under the Manufacturer Discount Program. In accordance with this appeals process, the manufacturer would have 60 calendar days after receipt of the civil monetary penalty notification to request a hearing (42 CFR 423.1020). If the manufacturer requests a hearing, the procedures outlined in section 1128A of the Act and operationalized by 42 CFR part 423, subpart T would apply. As set forth in section 1128A(f) of the Act, if the manufacturer does not pay the civil monetary penalty timely, the civil monetary penalty amount may be deducted from any sum then or later owing by the United States. Civil monetary penalty funds would be deposited in accordance with section 1128A(f) of the Act.
As described in proposed § 429.1020(e), in the event that a manufacturer declares bankruptcy, as described in title 11 of the United States Code and fails to pay either the full rebate amount of a Biosimilar Delay rebate owed or the total sum of civil monetary penalties imposed, the government reserves the right to file a proof of claim with the bankruptcy court to recover the unpaid rebate amount and/or civil monetary penalties owed by the manufacturer. Filing a proof of claim does not waive any other rights of the United States to recover such amounts or to assert priority status as permitted under applicable law. CMS may exercise its offset authority under section 1128A(f) of the Act independently of, and in addition to, other available collection remedies permitted under Federal law.
K. Application of Medicare Part B and Part D Drug Inflation Rebate Programs to Selected Drugs
Section 1847A(i) of the Act requires that manufacturers of Part B rebatable drugs pay inflation rebates to Medicare for certain Part B rebatable drugs based on specific requirements and formulas. Section 1860D-14B of the Act requires that manufacturers of Part D rebatable drugs pay inflation rebates to Medicare for certain Part D rebatable drugs based on specific requirements and formulas.
As described in section 120 of the Negotiation Program Guidance, whether a drug is a selected drug will have no bearing as to whether the drug is also subject to the Medicare Part B and Part D Drug Inflation Rebate Program. However, when a selected drug is no longer considered to be a selected drug, certain components of the applicable rebate amount formula are recalculated. As such, in the calendar year (CY) 2025 Physician Fee Schedule (PFS) final rule (89 FR 98582 through 98583), at § 427.303(c)(5) and (e)(5), we codified the identification of the payment amount benchmark quarter and the identification of the benchmark period CPI-U, respectively, in the case when a Part B rebatable drug is no longer considered to be a selected drug. Additionally, in the CY 2025 PFS final rule (89 FR 98591 through 98592), at § 428.202(c)(5) and (e)(5), we codified the identification of the payment amount benchmark period and the identification of the benchmark period CPI-U, respectively, in the case when a Part D rebatable drug is no longer considered to be a selected drug.
III. Proposed Implementation of Inflation Reduction Act Provisions for the Medicare Prescription Drug Benefit Program
A. Part D Formulary Inclusion of Selected Drugs
1. Background
Sections 11001 and 11002 of the Inflation Reduction Act of 2022 (IRA) (Pub. L. 117-169) created Part E under Title XI of the Act (sections 1191 through 1198) which established the Negotiation Program to negotiate maximum fair prices (MFPs) for certain high expenditure, single source drugs and biological products.
Section 11001(b) of the IRA added section 1860D-4(b)(3)(I)(i) of Act, which requires that, starting in 2026 and for each subsequent year, Part D plan sponsors include on their formularies each covered Part D drug that is a selected drug under section 1192 of the Act for which an MFP (as defined in section 1191(c)(3) of the Act) is in effect with respect to the year. Section 11001(b) of the IRA also added section 1860D-4(b)(3)(I)(ii) of the Act, which clarifies that nothing in clause (i) shall be construed as prohibiting Part D plan sponsors from removing from their formularies such a selected drug if such removal would be permitted under § 423.120(b)(5)(iv) or any successor regulation.
Section 11001(c) of the IRA directed the Secretary to implement the provisions in section 11001 of the IRA, including amendments made by such section, for 2026, 2027, and 2028, by program instruction or other forms of program guidance. In accordance with the law, CMS has issued several guidance documents for implementing the Negotiation Program, including the requirement that Part D plan sponsors include on their formularies selected drugs for which an MFP is in effect, starting in 2026.
CMS issued revised or final guidance on June 30, 2023, October 2, 2024, and September 30, 2025 for implementation of the Negotiation Program for initial price applicability years 2026, 2027, and 2028, respectively.[66]
In these guidance documents, CMS described the requirement for Part D plan sponsors to include on their formularies each covered Part D drug that is a selected drug under section 1192 of the Act for which an MFP (as defined in section 1191(c)(3) of the Act) is in effect with respect to the year as well as the exception that allows Part D plan sponsors to remove a selected drug from their formulary if such removal meets the requirements specified in § 423.120(b)(5)(iv) or any successor regulation. As discussed in section I.A.2, in this proposed rule, unless otherwise specified, references hereinafter to “the Negotiation Program Guidance” are to the most recent program guidance published by CMS, which is the Medicare Drug Price Negotiation Program: Final Guidance, Implementation of Sections 1191-1198 of the Social Security Act for Initial Price Applicability Year 2028 and Manufacturer Effectuation of the Maximum Fair Price in 2026, 2027, and 2028 that was published on September 30, 2025.
On April 7, 2025, CMS issued the Final CY 2026 Part D Redesign Program Instructions which described certain changes under the IRA to the Part D benefit for CY 2026.[67]
In section 90 of these program instructions, CMS identified § 423.120(e)(2)(i), and the corresponding notice requirements at § 423.120(f)(2), (3), and (4), as the successor regulation for purposes of implementing section 1860D-4(b)(3)(I)(ii) of the Act, as added by
( printed page 36318)
section 11001(b) of the IRA. In section 110.1 of the Negotiation Program Guidance, CMS incorporated section 90 of the Final CY 2026 Part D Redesign Program Instructions with respect to the successor regulation exception and extended the policies therein to 2027 and 2028.
In this proposed rule, we are proposing new § 423.120(b)(2)(vii) and (viii) to codify the requirement that Part D plan sponsors include each Part D drug that is a selected drug with an MFP in effect on their formularies and the exception that permits Part D plan sponsors to remove such a selected drug if the removal would be permitted under the successor regulation at § 423.120(e)(2)(i), (f)(2), (3), and (4) that we are proposing to codify. In alignment with these changes, we are also proposing to codify a conforming change to the definition of corresponding drug at § 423.100.
2. Part D Formulary Inclusion of Selected Drugs (§ 423.120)
Section 11001(b) of the IRA added section 1860D-4(b)(3)(I)(i) of the Act to require that for 2026 and each subsequent year, Part D plans include each covered Part D drug that is a selected drug under section 1192 of the Act on Part D formularies if an MFP is in effect for that drug with respect to that year. For 2026, 2027, and 2028, we have implemented this requirement through guidance.[68]
With the expiration of the IRA program instruction requirement for the Negotiation Program at the end of 2028, we now propose to codify this requirement at new § 423.120(b)(2)(vii) with respect to selected drugs for which an MFP is in effect in 2029 and subsequent years.
While section 1860D-4(b)(3)(I)(i) of the Act specifies that Part D plans must include on their Part D formularies covered Part D drugs that are selected drugs for which an MFP is in effect with respect to the year, it does not otherwise specify formulary requirements for tier placement or utilization management. CMS received comments [69]
expressing concerns that plans may hinder beneficiary access to selected drugs (for example, by applying utilization management requirements that are not based on medical appropriateness or placing selected drugs on less favorable tiers compared to non-selected drugs). In response to these comments, we stated [70]
that CMS agrees on the importance of ensuring meaningful beneficiary access to selected drugs and their MFPs and ensuring that plans do not engage in behavior that hinders access to selected drugs or non-selected drugs when medically appropriate. However, CMS also understands that not all selected drugs and drug classes will present Part D plan sponsors and their Pharmacy and Therapeutics Committees with the same formulary considerations, and the same formulary placement might not be warranted in all situations. In order to ensure meaningful beneficiary access to selected drugs and their MFPs, CMS would continue to use its comprehensive formulary review process to assess any practices that may undermine beneficiary access to selected drugs and ensure that Part D plan sponsors comply with existing statutory and regulatory restrictions on formulary design.
Sections 1860D-2(b)(2)(B) and 1860D-4(c)(1)(A) of the Act permit Part D plan sponsors to use formularies and tiered cost sharing in their benefit design, subject to certain limitations, and require them to have a cost-effective drug utilization management program that includes incentives to reduce costs when medically appropriate. Under section 1860D-11(e)(2)(D)(i) of the Act, CMS may approve a prescription drug plan only if the agency does not find that the design of the plan and its benefits (including any formulary and tiered formulary structure) are likely to substantially discourage enrollment by certain part D eligible individuals under the plan. In addition, § 423.272(b)(2)(i) requires that CMS not approve a bid if it finds that the design of the plan and its benefits (including any formulary and tiered formulary structure) or its utilization management program are likely to substantially discourage enrollment by certain Part D eligible individuals under the plan. Further, § 423.120(b)(2)(iii) requires each Part D plan formulary to “include adequate coverage of the types of drugs most commonly needed by Part D enrollees, as recognized in national treatment guidelines”. Finally, § 423.120(b)(1)(v) requires that in making decisions about formulary design, the entity designing the formulary must “base clinical decisions on the strength of scientific evidence and standards of practice”. CMS maintains a robust clinical formulary review process to ensure that all Medicare Part D plans meet these and other applicable requirements. CMS reviews all formularies annually to ensure that each formulary meets the agency's clinical review criteria, which include comprehensive evaluation of tier placement and all utilization management restrictions and criteria.
Consistent with section 110 of the Negotiation Program Guidance, given CMS' statutory obligation to monitor Medicare Part D plans' compliance with all applicable formulary requirements, we would continue to use our formulary review process to assess: (1) any instances where Part D plan sponsors place selected drugs on non-preferred tiers; (2) any instances where a selected drug is placed on a higher cost-sharing tier than non-selected brand drugs in the same class; (3) any instances where Part D plan sponsors require utilization of an alternative non-selected brand drug prior to a selected drug (that is, step therapy); or (4) any instances where Part D plan sponsors impose more restrictive utilization management (for example, step therapy and prior authorization) for a selected drug compared to a non-selected brand drug in the same class.
For this review, we would consider, consistent with section 110 of the Negotiation Program Guidance, class to mean the FDA Established Pharmacologic Class or other source that groups like drugs with similar mechanisms of action. Specifically, CMS would expect Part D plan sponsors to provide a reasonable justification to support their submitted plan benefit design and formulary design that includes any of the practices noted previously during the annual bid review process. This justification should address applicable clinical factors, such as clinical superiority, non-inferiority, or equivalence of the selected and non-selected drugs, as well as the plan design's compliance with applicable statutory and regulatory requirements (for example, the requirement to have a cost-effective drug utilization management program that bases decisions on the strength of the clinical evidence and standards of practice). CMS would evaluate these justifications for compliance with applicable statutory and regulatory requirements and would approve a Part D plan bid submitted by a Part D plan sponsor only if the plan
( printed page 36319)
benefit design and formulary design complies with those requirements.
As discussed in the Negotiation Program Guidance, CMS also is aware that there are concerns that Part D plan sponsors could broadly shift access with respect to a drug selected for negotiation after the drug has been announced as a selected drug, that is, in the contract year prior to the selected drug's MFP taking effect. To address these concerns, CMS would continue to monitor trends in formulary placement for selected drugs beginning after drugs are selected for an initial price applicability year. We believe this approach would continue to provide Part D plan sponsors with the flexibility to continue to manage costs through utilization management in a clinically appropriate manner, while allowing us to monitor practices that may undermine beneficiary access to selected drugs and potentially inform new requirements for future contract years.
Finally, because a selected drug includes all dosage forms and strengths to which the MFP applies, section 1860D-4(b)(3)(I)(i) of the Act requires that formularies include all such dosage forms and strengths of the selected drug that constitute a covered Part D drug and for which the MFP is in effect. Thus, consistent with section 110 of the Negotiation Program Guidance, we are proposing that Part D plan sponsors would continue to be required to include all such dosage forms and strengths of the selected drug that constitute a Part D drug and for which the MFP is in effect on their formularies.
In summary, in alignment with section 1860D-4(b)(3)(I)(i) of the Act, as added by section 11001(b) of the IRA, we are proposing to codify at new § 423.120(b)(2)(vii) the requirement that, for 2026 and each subsequent year, Part D plan sponsors include each Part D drug that is a selected drug under section 1192 of the Act for which a maximum fair price (as defined in section 1191(c)(3) of the Act) is in effect with respect to the year.
3. Successor Regulation Exception Permitting Formulary Substitutions of Selected Drugs
Section 11001(b) of the IRA added section 1860D-4(b)(3)(I)(ii) of the Act, which states that nothing in section 1860D-4(b)(3)(I)(i) of the Act shall be construed as prohibiting a Part D plan sponsors from removing a selected drug from a formulary if such removal would be permitted under § 423.120(b)(5)(iv) or any successor regulation.
At the time the IRA was enacted, then-current § 423.120(b)(5)(iv) permitted a plan to immediately substitute on the formulary a newly available generic drug for its brand name drug if certain notice and timing requirements were met. However, due to changes made to the regulations in the Contract Year 2025 Parts C/D Final Rule (89 FR 30448), there is no longer a § 423.120(b)(5)(iv) in the current Part D regulations. Under the current Part D regulations, the approval requirements for immediate substitutions, which were revised to provide for the substitution of additional types of products, are now codified at § 423.120(e)(2)(i), and the corresponding notice requirements for such formulary changes are now codified at § 423.120(f)(2), (3), and (4).
Because there was no longer a § 423.120(b)(5)(iv) in the Part D regulations, CMS had to identify the successor regulation to § 423.120(b)(5)(iv) for the purposes of the exception to the formulary inclusion requirement for selected drugs in section 1860D-4(b)(3)(I)(ii) of the Act. Because section 11001(c) of the IRA directed the Secretary to implement the provisions in section 11001 of the IRA, including amendments made by such section, for 2026, 2027, and 2028, by program instruction or other forms of program guidance, CMS identified the successor regulation for such years by guidance. For 2026, CMS identified the successor regulation in section 90 of the Final CY 2026 Part D Redesign Program Instructions. For 2027 and 2028, in section 110.1 of the Negotiation Program Guidance, CMS incorporated section 90 of the Final CY 2026 Part D Redesign Program Instructions with respect to the successor regulation exception and extended the policies therein to 2027 and 2028.
With the expiration of the IRA program instruction requirement for the Negotiation Program at the end of 2028, we now propose to codify, consistent with our previously issued guidance in the Final CY 2026 Part D Redesign Program Instructions and the Negotiation Program Guidance, the section 1860D-4(b)(3)(I)(ii) of the Act exception to the formulary inclusion requirement for selected drugs at new § 423.120(b)(2)(viii). We are also proposing, consistent with the Final CY 2026 Part D Redesign Program Instructions and the Negotiation Program Guidance, to codify our identification of § 423.120(e)(2)(i), (f)(2), (3), and (4) as the “successor regulation” for the purposes of implementing section 1860D-4(b)(3)(I)(ii) of the Act, and proposed § 423.120(b)(2)(viii). In alignment with these changes, we are also proposing to codify a conforming change to the definition of corresponding drug at § 423.100.
a. Exception Permitting Formulary Substitutions of Selected Drugs
As discussed in the previous section, we are proposing to codify our identification of § 423.120(e)(2)(i), (f)(2), (3), and (4) as the successor regulation for purposes of implementing section 1860D-4(b)(3)(I)(ii) of the Act. Under § 423.120(e)(2)(i), a Part D plan sponsor is permitted to—
[M]ake negative formulary changes to a brand name drug, a reference product, or a brand name biological product within 30 days of adding a corresponding drug to its formulary on the same or lower cost sharing tier and with the same or less restrictive formulary prior authorization (PA), step therapy (ST), or quantity limit (QL) requirements, so long as the Part D sponsor previously could not have included such corresponding drug on its formulary when it submitted its initial formulary for CMS approval . . . because such drug was not yet available on the market, and the Part D sponsor has provided advance general notice as specified in paragraph (f)(2) of [section 423.120].
Under § 423.120(f)(2), (3), and (4), a Part D plan sponsor making an immediate substitution under § 423.120(e)(2)(i) is required to provide advance general notice and retrospective notice to enrollees, among others, and to ensure that written notices include specified content. Specifically, the advance general notice must be provided to all current and prospective enrollees and other specified entities, must be in the Part D plan sponsor's formulary and other applicable beneficiary communication materials, and must advise that the Part D plan sponsor may make immediate negative formulary changes, consistent with the regulation, at any time. The required retrospective notice must be provided to affected enrollees as soon as possible, but no later than by the end of the month following any month in which the change takes effect. The content of these notices is specified in § 423.120(f)(4).
Under this successor regulation, nothing in section 1860D-4(b)(3)(I)(i) of the Act, and proposed § 423.120(b)(2)(vii), would be construed as prohibiting a Part D plan sponsor from removing a selected drug from its formulary if, in accordance with § 423.120(e)(2)(i) and the notice requirements of § 423.120(f)(2), (3), and (4), the Part D plan sponsor adds to its formulary on the same or lower cost sharing tier and with the same or less restrictive PA, ST, or QL requirements a newly available corresponding drug, as defined at § 423.100, with respect to such selected drug.
( printed page 36320)
By proposing to codify the current identification of § 423.120(e)(2)(i) as part of the successor regulation, we are also proposing to codify that, in addition to continuing to permit Part D plan sponsors to remove a selected drug that is a brand name drug and replace it with a generic drug as an immediate substitution, we would also continue to permit Part D plan sponsors to remove a selected drug that is a reference product and replace it with an interchangeable biological product as an immediate substitution.[71]
As discussed in section 90 of the Final CY 2026 Part D Redesign Program Instructions, allowing removal of selected drugs that are reference products and replacement with interchangeable biological products as immediate substitutions under the successor regulation that was first identified for 2026 was similar in kind to and consistent with the original regulation that was identified in the statute for the exception. Our proposed codification of the successor regulation in this rule would continue to apply the same rules to interchangeable biological products as are applied to generic drugs. As we explained when we first identified the successor regulation for 2026, this promotes consistency across selected drugs because, regardless of whether a selected drug is a drug or biological product, the Part D plan sponsor is able to remove a selected drug that is a brand name drug or reference product as an immediate substitution.
CMS would continue to permit, consistent with the agency's longstanding practice, including under § 423.120(b)(5)(iv) at the time of enactment of the IRA and under the identified successor regulation for 2026, 2027, and 2028, Part D plan sponsors to immediately substitute a selected drug for which there is a generic drug or interchangeable biological product available in the same dosage form, route of administration, and strength. The exception to the selected drug formulary inclusion requirement in section 1860D-4(b)(3)(I)(ii) of the Act allows for a selected drug to be removed as permitted under § 423.120(b)(5)(iv) (or any successor regulation). Accordingly, it is consistent with the exception established under section 1860D-4(b)(3)(I)(ii) of the Act to continue to apply this longstanding policy under the Part D program to the removal of selected drugs under the proposed codification of the current successor regulation pursuant to section 1860D-4(b)(3)(I)(ii) of the Act, and proposed § 423.120(b)(2)(viii).
Consistent with § 423.120(e)(2)(i), Part D plan sponsors that implement an immediate substitution for any brand name drug or reference product that is a selected drug on their formulary in accordance with section 1860D-4(b)(3)(I)(ii) of the Act, and proposed § 423.120(b)(2)(viii), would not be required to exempt enrollees who were already taking the selected drug. An enrollee may avail themselves of the formulary exception process, in accordance with § 423.578(b), if it is medically necessary for them to remain on a selected drug that has been removed from the formulary.
As discussed in the comment responses of the Final CY 2026 Part D Redesign Program Instructions, we also note that Part D plan sponsors would not need to remove selected drugs from their formularies to add a new generic drug, interchangeable biological product, or biosimilar biological product other than an interchangeable biological product to their formularies. A Part D plan sponsor can make such an addition at any time during the plan year, while keeping the brand name drug or reference product on the formulary. In addition, we remind Part D plan sponsors that they may also make maintenance changes with respect to selected drugs other than removing them from the formulary, in accordance with § 423.120(e) and the notice requirements of § 423.120(f).
In the Draft CY 2026 Part D Redesign Program Instructions,[72]
CMS solicited comments on alternative approaches to identifying the successor regulation. These proposed alternative approaches included identifying additional regulations to expand the successor regulation exception to encompass maintenance changes, in addition to immediate substitutions. While we maintain that section 1860D-4(b)(3)(I)(ii) of the Act gives CMS the authority to identify such maintenance changes as part of the successor regulation for purposes of section 1860D-4(b)(3)(I)(ii) of the Act, CMS continues to decline to do so at this time and is proposing to maintain § 423.120(e)(2)(i), (f)(2), (3), and (4) as the successor regulation. However, in the future, CMS may identify new regulations that constitute the successor regulation (for example, as the biosimilar market matures or as additional changes are made to the underlying regulations).
In summary, consistent with the Negotiation Program Guidance and the Final CY 2026 Part D Redesign Program Instructions, CMS is proposing to codify the identification of § 423.120(e)(2)(i), (f)(2), (3), and (4) as the successor regulation for the purposes of section 1860D-4(b)(3)(I)(ii) of the Act and add a new paragraph at § 423.120(b)(2)(viii) that specifies that nothing shall prohibit a Part D plan sponsor from removing a selected drug from their formulary if such removal meets the requirements specified in § 423.120(e)(2)(i) and the notice requirements specified in paragraphs (f)(2), (3), and (4) of this section.
b. Corresponding Drugs Do Not Include Selected Drugs
Consistent with CMS' proposed codification of the current identification of § 423.120(e)(2)(i), (f)(2), (3), and (4) as the successor regulation for the purposes of section 1860D-4(b)(3)(I)(ii) of the Act, a Part D plan sponsor may remove a selected drug from its formulary if, in accordance with § 423.120(e)(2)(i) and the notice requirements of § 423.120(f)(2), (3), and (4), the Part D plan sponsor adds to its formulary on the same or lower cost sharing tier and with the same or less restrictive PA, ST, or QL requirements a newly available corresponding drug with respect to such selected drug.
Section 423.100 currently defines “corresponding drug” as “respectively, a generic or authorized generic of a brand name drug, an interchangeable biological product of a reference product, or an unbranded biological product marketed under the same biologics license application (BLA) as a brand name biological product.” As discussed in section 90 of the Final CY 2026 Part D Redesign Program Instructions, when read in isolation, the current definition might incorrectly appear to suggest that a Part D plan sponsor could remove a selected drug under section 1860D-4(b)(3)(I)(ii) of the Act, if the Part D plan sponsor adds an authorized generic of the brand name drug or an unbranded biological product marketed under the same BLA as the brand name biological product.
( printed page 36321)
However, such a removal would be inconsistent with the Part D plan sponsor's obligation under section 1860D-4(b)(3)(I)(i) of the Act, and proposed § 423.120(b)(2)(vii), to include on its formulary each covered Part D drug that is a selected drug under section 1192 of the Act for which an MFP is in effect with respect to the year.
As stated in section 1192(e)(2)(A) of the Act (as added by section 11001 of the IRA), and as discussed in section II.B.6 of this proposed rule, an authorized generic drug and the qualifying single source drug that is the listed drug or reference product of that authorized generic drug shall be treated as the same qualifying single source drug and, thus, the same selected drug. For the purposes of the Negotiation Program, an “authorized generic drug” is defined in section 1192(e)(2)(B) of the Act and in proposed § 429.20 as:
(1) in the case of a drug product, an authorized generic drug (as such term is defined in section 505(t)(3) of the FD&C Act); and
(2) in the case of a biological product, a product that has been licensed under section 351(a) of the PHS Act and is marketed, sold, or distributed directly or indirectly to the retail class of trade under a different labeling, packaging (other than repackaging as the reference product in blister packs, unit doses, or similar packaging for institutions), product code, labeler code, trade name, or trademark than the reference product.
As discussed in section II.B.6.a of this proposed rule, authorized generics of a brand name drug that is a selected drug and unbranded biological products marketed under the same license as a brand name biological product that is a selected drug are treated as the same selected drug as the respective listed drug or reference product.
For example, section II.B.6.a of this proposed rule states that, for drug products, CMS is proposing at § 429.125(b)(1) to identify a potential qualifying single source drug using all dosage forms and strengths of the drug with the same active moiety and the same holder of a New Drug Application (NDA), inclusive of products that are marketed under different NDAs. The potential qualifying single source drug also includes all dosage forms and strengths of the drug with the same active moiety and marketed under the same NDA(s) that are authorized generic drugs (defined in section 1192(e)(2)(B)(i) of the Act and at proposed § 429.20) that are marketed under such NDA(s). Likewise, section II.B.6.a of the proposed rule states that, for biological products, CMS is proposing at § 429.125(b)(2) to identify a potential qualifying single source drug using all dosage forms and strengths of the biological product with the same active ingredient and the same holder of a Biologics License Application (BLA), inclusive of products that are marketed under different BLAs. The potential qualifying single source drug also includes all dosage forms and strengths of the biological product with the same active ingredient and marketed under the same BLA(s) that are authorized generic drugs, the definition of which at section 1192(e)(2)(B)(ii) of the Act and proposed § 429.20 includes unbranded biological products that are marketed under such BLA(s).
Because an authorized generic of a brand name drug that is a selected drug or an unbranded biological product marketed under the same BLA as a brand name biological product that is a selected drug also qualifies as the selected drug, section 1860D-4(b)(3)(I)(i) of the Act, and proposed § 423.120(b)(2)(vii), requires the Part D plan sponsor to include each such authorized generic or unbranded biological product that is a covered Part D drug for which an MFP is in effect on its formulary.
Consequently, the statute does not permit a Part D plan sponsor to remove a selected drug that is a brand name drug or brand name biological product on the basis of adding an authorized generic of the brand name drug or an unbranded biological product marketed under the same BLA as the brand name biological product.
In situations where a selected drug includes both a brand name biological product and an unbranded biological product marketed under the same BLA as the brand name biological product, a Part D plan sponsor may remove both the brand name biological product and the unbranded biological product from its formulary when making an immediate substitution under § 423.120(e)(2)(i) by adding a single interchangeable biological product to its formulary. The interchangeable biological product that is the corresponding drug for the selected drug may replace both the reference product and the unbranded biological product provided that the requirements specified in the proposed successor regulation at § 423.120(e)(2)(i), (f)(2), (3), and (4) are met.
To ensure consistency with the IRA's formulary inclusion requirement, which we are proposing to codify at § 423.120(b)(2)(vii), and avoid any potential confusion related to the identification of § 423.120(e)(2)(i) as part of the successor regulation to § 423.120(b)(5)(iv) for the purposes of section 1860D-4(b)(3)(I)(ii) of the Act, we amended the definition of “corresponding drug” in section 90 of the Final CY 2026 Part D Redesign Program Instructions, consistent with the statutory directive to implement section 11001 of the IRA, including amendments made by such section, by program instruction or other forms of program guidance. Specifically, section 90 of such program instructions defined “corresponding drug” for 2026 to “mean[ ], respectively, a generic or authorized generic of a brand name drug, an interchangeable biological product of a reference product, or an unbranded biological product marketed under the same biologics license application (BLA) as a brand name biological product. A corresponding drug does not include a selected drug as defined in section 1192(c) of the Act.” Section 110.1 of the Negotiation Program Guidance incorporated section 90 of the Final CY 2026 Part D Redesign Program Instructions with respect to the successor regulation exception and extended the policies therein, including the revised definition of “corresponding drug,” to 2027 and 2028.
With the expiration of the IRA program instruction requirement for the Negotiation Program at the end of 2028, CMS is proposing to codify for 2029 and subsequent years the current definition set forth in guidance at § 423.100. Specifically, the definition at § 423.100 would include language stating that a corresponding drug does not include a selected drug, as defined in section 1192(c) of the Act.
c. Timing of Immediate Substitutions
Section 423.120(e)(2)(i), (f)(2), (3), and (4), which we are proposing to codify as the successor regulation consistent with current guidance, permit a Part D plan sponsor, provided it has met the notice requirements, to remove a selected drug that is a brand name drug or reference product from its formulary and replace it with a generic of the brand name drug or an interchangeable biological product of the reference product “so long as the Part D sponsor previously could not have included such corresponding drug on its formulary
when it submitted its initial formulary
for CMS approval consistent with paragraph (b)(2) of this section because such drug was
not yet available on the market
” [emphasis added]. As such, under these regulations, for Part D plan sponsors that have a selected drug for initial price applicability year 2029 on their 2028 formulary, if a generic drug or interchangeable biological product of a selected drug becomes available on the market in 2028 after the Part D plan
( printed page 36322)
sponsor submitted its initial 2029 formulary for CMS approval, consistent with our longstanding policy on immediate substitutions, a Part D plan sponsor could add such generic drug or interchangeable biological product and remove the selected drug from its formulary as an immediate substitution for 2028, as well as for 2029. In other words, where the Part D plan sponsor could not have included the generic drug or interchangeable biological product on either its 2028 initial formulary submission or its 2029 initial formulary submission, and such Part D plan sponsor removes the selected drug as an immediate substitution, the Part D plan sponsor can apply the removal to both the current year formulary as well as the already-submitted formulary for the following year.
A Part D plan sponsor that does not have the selected drug on its 2028 formulary, but has submitted a formulary for 2029 that includes the selected drug, in accordance with section 1860D-4(b)(3)(I)(i) of the Act, could remove the selected drug as part of an immediate substitution only with respect to its 2029 formulary. That is, if a generic drug of a brand name drug that is a selected drug or an interchangeable biological product of a reference product that is a selected drug was first available on the market after the initial submission of the 2029 formulary, the Part D plan sponsor could, in the latter part of 2028, remove that selected drug from its 2029 formulary.
This application of the immediate substitutions policy to both the 2028 and 2029 formularies with respect to a generic drug or interchangeable biological product that becomes available on the market after the Part D plan sponsor has already submitted its initial formulary to CMS is consistent with section 1860D-4(b)(3)(I) of the Act. As discussed previously, the formulary inclusion requirement in section 1860D-4(b)(3)(I) of the Act applies with respect to selected drugs starting in initial price applicability year 2026 and the only permitted exception to the requirement is the “removal” of “such selected drug.” The plain text of the statute contemplates that a selected drug will need to be included on the plan's formulary for the first initial price applicability year in which the MFP for the selected drug is in effect, regardless of whether the plan ever previously included the drug. Moreover, the only statutory exception to the formulary inclusion obligation is the “removal” of “such a selected drug” in accordance with § 423.120(b)(5)(iv) or its successor regulation, which we are proposing to codify as § 423.120(e)(2)(i), (f)(2), (3), and (4). The references to “removal” and “such selected drug” indicate that the selected drug must first be included on the formulary for the initial price applicability year under section 1860D-4(b)(3)(I)(i) of the Act and only then can be removed under section 1860D-4(b)(3)(I)(ii) of the Act.
Accordingly, as discussed previously, a Part D plan sponsor that includes a selected drug with an initial price applicability year of 2029 on its initial formulary submission for 2029 could remove the selected drug as part of an immediate substitution prior to the start of 2029 if a generic drug or interchangeable biological product of the selected drug becomes available on the market in 2028 after the initial formulary submission. In such cases, the Part D plan sponsor will have included the selected drug on the formulary with respect to the initial price applicability year in which the MFP is in effect (that is, 2029), as required by section 1860D-4(b)(3)(I)(i) of the Act, and subsequently removed the selected drug, as permitted by section 1860D-4(b)(3)(I)(ii) of the Act.
Further, we note that there may be scenarios in which a generic drug or interchangeable biological product of, for example, a selected drug with an initial price applicability year of 2029 becomes available on the market in 2028 (after the Part D plan sponsor has submitted its initial 2028 formulary in 2027) but before the Part D plan sponsor submits its 2029 initial formulary for CMS approval in 2028. Under such a scenario, a Part D plan sponsor would be permitted, assuming all requirements are met, to remove the selected drug with an initial price applicability year of 2029 (prior to its MFP taking effect) from its 2028 formulary under § 423.120(e)(2)(i), but would be required under section 1860D-4(b)(3)(I)(i) of the Act to include the selected drug with an initial price applicability year of 2029 on its 2029 formulary when the MFP takes effect.
Consistent with section 90 of the Final CY 2026 Part D Redesign Program Instructions, if there is a generic drug or interchangeable biological product for a selected drug with an initial price applicability year of 2029 and such generic drug or interchangeable biological product is available on the market before a Part D plan sponsor's 2029 initial formulary submission, such Part D plan sponsor would still need to include the selected drug on its 2029 formulary submission, regardless of whether the Part D plan sponsor had removed such selected drug from its 2028 formulary via an immediate substitution in 2028, to comply with the formulary inclusion requirement in section 1860D-4(b)(3)(I)(i) of the Act. Moreover, the Part D plan sponsor would not be permitted to remove the selected drug from the formulary for 2029 as an immediate substitution and replace it with the generic drug or interchangeable biological product that became available on the market prior to the initial formulary submission for 2029 because section 1860D-4(b)(3)(I)(ii) of the Act permits removal only in accordance with § 423.120(b)(5)(iv) or its successor regulation. As discussed previously, the successor regulation, which we propose to codify for 2029 and subsequent years, includes § 423.120(e)(2)(i), which, like § 423.120(b)(5)(iv) at the time of enactment, permits an immediate substitution only if the Part D plan sponsor previously could not have included such corresponding drug on its formulary when it submitted its initial formulary for CMS approval. In this scenario, because the Part D plan sponsor could have included the generic drug or interchangeable biological product in its initial formulary submission for 2029, an immediate substitution would not be available.
Further, consistent with section 90 of the Final CY 2026 Part D Redesign Program Instructions, the regulatory language at § 423.120(e)(2)(i), that a Part D plan sponsor previously could not have included the corresponding drug on its formulary when it submitted its initial formulary for CMS approval, would continue to mean, in practice, that the corresponding drug is not included on the final formulary reference file (FRF) update that CMS releases before the bid submission deadline. Consistent with our longstanding practice, the determination of whether a new generic drug or interchangeable biological product is included on the final FRF update that CMS releases before the bid submission deadline will be based on the presence of a RxNorm Concept Unique Identifier (RxCUI) on that FRF update that represents the generic drug or interchangeable biological product.
Finally, consistent with section 110.1 of the Negotiation Program Guidance, we are proposing that removals under the statutory formulary inclusion exception, which we are proposing to codify at new § 423.120(b)(2)(viii), cannot be carried over to subsequent years within the price applicability period simply because a selected drug was removed in a preceding year during the price applicability period. Instead, any removal must independently meet the immediate substitution requirements for each plan year
( printed page 36323)
because, consistent with CMS' longstanding policy, CMS considers each plan year's formulary to be separate and distinct from the prior year. Therefore, a removal could only apply to multiple plan years in a price applicability period if it independently meets the immediate substitution requirements for each applicable plan year.
B. Negotiated Price for Selected Drugs
Section 1860D-2(d)(1) of the Act requires Part D sponsors to provide beneficiaries with access to negotiated prices for covered Part D drugs. Section 1860D-2(d)(1)(A) of the Act requires Part D sponsors to “provide enrollees with access to negotiated prices used for payment for covered Part D drugs.” Subparagraph (B) further clarifies that negotiated prices, subject to subparagraph (D), “shall take into account negotiated price concessions, such as discounts, direct or indirect subsidies, rebates, and direct or indirect remunerations, for covered Part D drugs, and include any dispensing fees for such drugs.”
The negotiated price is the price paid to the network pharmacy or other network dispensing provider for a covered Part D drug dispensed to a plan enrollee and reported to CMS at the point of sale by the Part D sponsor. This point of sale price is used to calculate beneficiary cost sharing. The negotiated price also serves as the primary basis for adjudicating the Part D benefit because it is used to determine plan, beneficiary, manufacturer, and government liability during the payment year, subject to final reconciliation after the coverage year ends.
In our final rule, “Medicare Program; Medicare Prescription Drug Benefit,” which appeared in the January 28, 2005
Federal Register
(70 FR 4194) (hereinafter referred to as the January 2005 final rule), we first codified the definition of negotiated prices described at section 1860D-2(d)(1) of the Act at § 423.100. We explained that the Act required “negotiated prices . . . to take into account negotiated price concessions for covered Part D drugs such as discounts, direct or indirect subsidies, rebates, and direct or indirect remunerations, and would include any applicable dispensing fees.” [73]
Accordingly, we defined negotiated prices at § 423.100 to mean prices for covered Part D drugs that—
Are available to beneficiaries at the point of sale at network pharmacies;
Are reduced by those discounts, direct or indirect subsidies, rebates, other price concessions, and direct or indirect remunerations that the Part D sponsor has elected to pass through to Part D enrollees at the point of sale; and
Includes any dispensing fees.
After issuing the January 2005 final rule, we revised the definition of negotiated prices at § 423.100 several times. Most significantly, in the final rule “Medicare Program; Contract Year 2015 Policy and Technical Changes to the Medicare Advantage and the Medicare Prescription Drug Benefit Programs,” which appeared in the May 23, 2014
Federal Register
(79 FR 29844) we amended the definition of “negotiated prices” at § 423.100 to require Part D sponsors to include all pharmacy price concessions and incentive payments to pharmacies in the negotiated price at the point of sale. The rule also established an exception that allowed sponsors to exclude contingent pharmacy payment adjustments that cannot reasonably be determined at the point of sale (the reasonably determined exception).
In the May 9, 2022
Federal Register
, we further amended § 423.100 by revising the term “negotiated prices” (plural) to “negotiated price” (singular) to clarify that a negotiated price may be set for each covered Part D drug. Based on feedback from stakeholders and information submitted by plan sponsors in their annual direct and indirect remuneration (DIR) reports indicating that sponsors had applied the reasonably determined exception more broadly than initially envisioned, we defined “negotiated price” as the lowest possible reimbursement a network pharmacy receives, in total, for a particular drug, taking into account pharmacy price concessions, and thus eliminated the reasonably determined exception. As a result, the definition of “negotiated price” at § 423.100 currently means the price for a covered Part D drug that—
The Part D sponsor (or other intermediary contracting organization) and the network dispensing pharmacy or other network dispensing provider have negotiated as the lowest possible reimbursement such network entity will receive, in total, for a particular drug;
Meets all of the following:
++ Includes all price concessions (as defined in this section) from network pharmacies or other network providers.
++ Includes any dispensing fees.
++ Excludes additional contingent amounts, such as incentive fees, if these amounts increase prices; and
Is reduced by non-pharmacy price concessions and other direct or indirect remuneration that the Part D sponsor passes through to Part D enrollees at the point of sale.
Section 11001(b) of the IRA amended section 1860D-2(d)(1) of the Act by adding subparagraph (D). That provision requires that, in the case of a covered Part D drug that is a selected drug, with respect to a price applicability period, the negotiated price “used for payment (as described in this subsection) shall be no greater than the [maximum fair price] for such drug . . . plus any dispensing fees for such drug.” Section 11001(b) of the IRA also added “subject to subparagraph (D)” to section 1860D-2(d)(1)(B) of the Act.
Section 11001(c) of the IRA directed CMS to implement section 11001 of the IRA, including the amendments made by such section, by program instruction or other forms of program guidance for initial price applicability years 2026 through 2028. Accordingly, we did not revise the regulatory definition of negotiated price to reflect the statutory amendment to section 1860D-2(d)(1) of the Act for purposes of 2026, 2027, and 2028. Instead, we applied the statutory negotiated price requirements for selected drugs through the applicable Negotiation Program guidance.
Most recently, in section 40.4 of the Negotiation Program Guidance, we stated that under section 1860D-2(d)(1)(D), the negotiated price for selected drugs “must not exceed the applicable [maximum fair price (MFP)] plus any dispensing fees for such drug.” [74]
CMS also stated that the statute requires manufacturers to provide access to the MFP for selected drugs to pharmacies, mail order services, and other dispensing entities with respect to MFP-eligible individuals who are dispensed such drugs.
With the expiration of the IRA program instruction requirement for the Negotiation Program at the end of 2028, we are proposing to codify the statutory amendments to the definition of negotiated price for selected drugs in the definition of “negotiated price” at § 423.100. The proposed revisions to § 423.100 would take effect with respect to 2029 and subsequent years.
Specifically, we propose to revise paragraph (2) of the definition to state that the required elements of the negotiated price are subject to a new paragraph (4) and add a new paragraph (4) to the definition to require that, for a covered Part D drug that is a selected
( printed page 36324)
drug, and for each year of a price applicability period with respect to such selected drug, the negotiated price used for payment, as described in paragraphs (1) through (3) of the negotiated price definition, must not exceed the MFP for such drug, plus any applicable dispensing fees.
In addition, we propose a revision to the regulatory definition of negotiated prices to codify longstanding requirements in guidance. Specifically, we have historically interpreted the definition of “negotiated price” under section 1860D-2(d)(1) of the Act to include sales tax. CMS currently requires Part D plan sponsors to report sales tax as part of the negotiated price on Prescription Drug Event (PDE) records.[75]
CMS has also historically interpreted and required the negotiated price to include vaccine administration fees, because beneficiaries do not purchase vaccines without the expectation that the vaccine will be administered.[76]
To better align the definition of “negotiated price” at § 423.100 with this historical interpretation, we propose to revise the required elements of the negotiated price in subparagraph (2) of the definition. Specifically, the word “and:” would be deleted from the end of paragraph (2)(ii), current paragraph (2)(iii) would be redesignated as paragraph (2)(v), a new paragraph (2)(iii) would be added to require the inclusion of sales tax, and a new paragraph (2)(iv) would be added to require the inclusion of vaccine administration fees. These changes would have the effect of adding sales tax and applicable administrative fees to the definition of “negotiated price” without altering any existing elements of that definition.
IV. Collection of Information Requirements
Under the Paperwork Reduction Act of 1995 (PRA) (44 U.S.C. 3501et seq.), we are required to provide notice in the
Federal Register
and solicit public comment before a “collection of information” requirement (as defined under 5 CFR 1320.3(c) of the PRA's implementing regulations) is submitted to the Office of Management and Budget (OMB) for review and approval. To fairly evaluate whether a collection of information should be approved by OMB, section 3506(c)(2)(A) of the PRA requires that we solicit comment on the following issues:
The need for information collection and its usefulness in carrying out the proper functions of our agency.
The accuracy of our estimate of the information collection burden.
The quality, utility, and clarity of the information to be collected.
Recommendations to minimize the information collection burden on the affected public, including automated collection techniques.
We are soliciting public comments (see section II. of this proposed rule) on each of the aforementioned issues for the following sections of this document that contain information collection requirements (ICRs). Comments, if received, will be responded to within the subsequent final rule.
A. Wage Estimates
To derive average costs, we used data from the U.S. Bureau of Labor Statistics' (BLS) May 2025 National Industry-Specific Occupational Employment and Wage Estimates for the Pharmaceutical and Medicine Manufacturing industry, when available, to derive average labor costs for all salary estimates.[77]
When industry-specific wage estimates were not available, the BLS' May 2025 Occupational Employment and Wage Statistics data was used (
https://www.bls.gov/oes/tables.htm). In this regard, Table 5 presents BLS' hourly median wage, our estimated cost of fringe benefits and other indirect costs (calculated at 100 percent of salary), and our adjusted hourly wage. There are many sources of variance in the average cost estimates, both because fringe benefits and other indirect costs vary significantly from employer to employer, and because methods of estimating these costs vary widely from study to study. Therefore, we believe that doubling the hourly median wage to estimate total cost is a reasonably accurate estimation method.
Table 5—BLS' Occupational Employment and Wage Estimates
Occupation title
Occupation code
Hourly
median wage
($/hr)
Fringe
benefits and
other indirect costs
($/hr)
Adjusted
hourly wage
($/hr)
All occupations 78
00-0000
24.51
24.51
49.02
Business Operations Specialists
13-1000
47.53
47.53
95.06
Chief Executive
11-1011
177.65
177.65
355.30
Cost Estimator
13-1051
41.41
41.41
82.82
Economist 79
19-3011
59.96
59.96
119.92
Financial Manager
11-3031
87.99
87.99
175.98
General and Operations Managers
11-1021
82.99
82.99
165.98
General Internal Medicine Physicians 80
29-1216
123.35
123.35
246.70
Lawyer
23-1011
142.57
142.57
285.14
Pharmacist
29-1051
66.73
66.73
133.46
Registered Nurse
29-1141
35.57
35.57
71.14
( printed page 36325)
B. Information Collection Requirements (ICRs)
1. ICRs Regarding the Drug Price Negotiation Program Under Sections 11001 and 11002 of the Inflation Reduction Act (CMS-10844, OMB 0938-1443 and CMS-10849, OMB 1948-1452) (§§ 429.110, 429.200, 429.300, 429.405, 429.445, 429.505, and 429.600 through 429.615)
a. Negotiation Program Drug Selection for Initial Price Applicability Year 20XX (§§ 429.110, 429.600 through 429.615)
The following was submitted to OMB for review under control number 0938-1443 (CMS-10844).
(1) Biosimilar Delay
This ICR addresses information for CMS to determine the applicability of section 1192(f)(1)(B) of the Act (which is proposed at § 429.110 of this proposed rule). Using information provided through this information collection in its determination, CMS may delay the inclusion of a negotiation-eligible drug that includes the reference product for a biosimilar biological product on the selected drug list for a given initial price applicability year if certain statutory requirements are met regarding the biosimilar's status of licensure and marketing (the “Biosimilar Delay”) in accordance with section 1192(f) of the Act.
CMS estimates collecting a total of 10 requests for an Initial Delay Request (defined in proposed § 429.20 and discussed in proposed § 429.110(b)) per year. We believe that collection of these data will be a one-time cost for each Biosimilar Manufacturer (defined in proposed § 429.20) for each negotiation-eligible drug for which it is seeking the Initial Delay Request for each initial price applicability year.
Using the wage rates in Table 5 of this proposed rule, we expect, for a Biosimilar Manufacturer, a lawyer 20.5 hours to gather and review the relevant ICR provisions, identify any controlled group members, identify and review any agreements between the Reference Manufacturer (defined in proposed § 429.20) and the Biosimilar Manufacturer, identify and review FDA licensure documentation and manufacturing schedule, trade agreements, and Securities and Exchange disclosures related to the Biosimilar Drug for each submission, and request technical assistance from CMS, a general and operations manager 4.5 hours to examine the gathered information, submit the form to CMS, and request technical assistance from CMS, and a chief executive 1.0 hour to review the information prior to submission and log into CMS' existing information technology system to certify the submission.
In aggregate, we estimated an annual burden of 260 hours (10 Biosimilar Manufacturers × 26.0 hr/response × 1 response/year) at a cost of $69,475.80 (10 responses × [(20.5 hr × $285.14/hr + (4.5 hr × $165.98/hr) + (1.0 hr × $355.30/hr)] (see Table 6).
Table 6—Summary of Total Annual Burden for Biosimilar Manufacturers To Complete an Initial Delay Request ICR Form
Requirement
Number of respondents
Total annual responses
Time per
response
(hours)
Total
annual time
(hours)
Labor
cost
($/hr)
Total cost
($)
Biosimilar Delay
10 Biosimilar Manufacturers
10 Biosimilar Manufacturers
26.0
260
Varies
69,475.80
(2) Identification and Selection of Renegotiation-Eligible Drugs
This ICR also offers Primary Manufacturers the voluntary option to submit information to CMS to inform CMS' determinations of which selected drugs qualify as a renegotiation-eligible drug, in accordance with section 1194(f)(2) of the Act, and as proposed at § 429.605 of this proposed rule, and may be selected for renegotiation in accordance with section 1194(f)(3) of the Act, and as proposed at § 429.610 of this proposed rule. Specifically, section 1194(f)(2)(D) of the Act provides that a selected drug is eligible for renegotiation if a new indication has been added to the selected drug or if CMS determines that there has been a material change to any of the factors listed in section 1194(e) of the Act and proposed at § 429.610(b) of this proposed rule.
CMS estimates collecting up to 36 responses for the burden estimate base year of initial price applicability year 2029 in response to the Identification and Selection of Renegotiation-Eligible Drugs ICR Form. This estimate of potential burden incorporates assumptions that a negotiated MFP will be agreed to for all selected drugs for initial price applicability year 2028 and that, at the time of this information collection, certain previously selected drugs will no longer be considered a selected drug by CMS consistent with section 1193(c) of the Act. This would include responses from all Primary Manufacturers of selected drugs with agreed-upon MFPs for prior initial price applicability years unless the selected drug has a change in monopoly status or a previously selected drug is no longer considered a selected drug by CMS consistent with section 1193(c) of the Act. The collection of these data will be a one-time cost for each selected drug and CMS assumes each Primary Manufacturer will spend, on average, the same amount of time to collect, aggregate, analyze, and report the data for a selected drug.
Using the wage rates in Table 5 of this proposed rule, we expect, for a Primary Manufacturer, a business operations specialist or team of business operations specialists 25.00 hours to gather cost data and compile required information, as specified in the data elements instructions, an economist or team of economists 75.00 hours to perform necessary economic analyses of data elements specified in the data element instructions, a financial manager 6.25 hours to review the results of all the analyses and cost estimates prior to submission to CMS, a lawyer 0.50 hours to review the compiled data submission, a cost estimator 17.75 hours to compile and report the required data to CMS, per the data element form instructions, and a chief executive 0.50 hours to review the data submission and log in to the CMS HPMS to certify the submission.
In aggregate, we estimated an annual burden of 4,500 hours (36 Primary Manufacturers × 125.00 hr/response × 1 response/year) at a cost of $513,383.40 (36 responses × [(25.00 hr × $95.06/hr + (75.00 hr × $119.92/hr) + (6.25 hr × $175.98/hr) + (0.50 hr × $285.14/hr) + (17.75 hr × $82.82/hr) + (0.50 hr × $355.30/hr)].
( printed page 36326)
Table 7—Summary of Total Annual Burden for Identification and Selection of Renegotiation-Eligible Drugs ICR Form
Requirement
Number of
respondents
Total annual responses
Time per
response
(hours)
Total
annual time
(hours)
Labor
cost
($/hr)
Total cost
($)
Identification and Selection of Renegotiation-Eligible Drugs
36 Primary Manufacturers
36 Primary Manufacturers
125.00
4,500
Varies
513,383.40
Table 8—Summary of TotaL Annual Burden for Negotiation Program Drug Selection ICR
Negotiation Program Drug Selection for Initial Price Applicability Year 20XX under Sections 11001 and 11002 of the Inflation Reduction Act (with base year of initial price applicability year 2029)
0938-1443 (CMS-10844)
46
46
Varies
4,760
Varies
582,859.20
b. Drug Price Negotiation for Initial Price Applicability Year 20XX (§§ 429.300, 429.405, 429.445, 429.505, 429.600, and 429.615)
The following was submitted to OMB for review under control number 0938-1452 (CMS-10849).
(1) Negotiation Data Elements for Selected Drugs for Negotiation From Primary Manufacturers
In accordance with section 1193(a)(4) and section 1194(b)(2)(A) of the Act and as proposed at § 429.200(b)(5) of this proposed rule, the Primary Manufacturer of a selected drug must submit, in a form and manner specified by CMS, information on the non-Federal average manufacturer price (“non-FAMP”) as defined in 38 U.S.C. 8126(h)(5) for the selected drug and information that CMS requires to carry out the negotiation process, including the factors outlined in section 1194(e)(1) of the Act, which, in conjunction with the available evidence on the factors outlined in section 1194(e)(2), will serve as the basis for determining the initial offer, any offer(s) associated with negotiation meeting(s), and the final offer, if applicable. In addition, manufacturers and the public may submit information on the factors outlined in section 1194(e)(2) of the Act, which describes evidence about the selected drug and its therapeutic alternative(s). This ICR serves as a way for a Primary Manufacturer of a selected drug to submit the required 1194(e)(1) data as well as serving as one of multiple ways that CMS will collect data described in section 1194(e)(2) of the Act. Although information submission for factors outlined in section 1194(e)(2) of the Act are voluntary and open to all interested parties, this burden estimate assumes that all Primary Manufacturers would choose to submit this type of data.
CMS estimates collecting up to 20 responses for burden estimate base year initial price applicability year 2029 from Primary Manufacturers. For purposes of this collection of information, this represents one response for each selected drug, and up to 20 drugs selected for negotiation, which represents the statutory maximum. The collection of these data will be a one-time cost for each selected drug and CMS assumes each Primary Manufacturer will spend, on average, the same amount of time to collect, aggregate, analyze, and report the data for a selected drug.
Using the wage rates in Table 5 of this proposed rule, we expect, for a Primary Manufacturer, a business operations specialist or team of business operations specialists 200 hours to gather cost data and compile required information, as specified in the data elements instructions, an economist or team of economists 600 hours to perform necessary economic analyses of data elements specified in the data element instructions, a financial manager 50 hours to review the results of all the analyses and cost estimates prior to submission to CMS, a lawyer 4 hours to review the compiled data submission, a cost estimator 142 hours to compile and report the required data to CMS, per the data element form instructions, and a chief executive 4 hours to review the data submission and log in to the CMS HPMS to certify the submission.
In aggregate, we estimated an annual burden of 20,000 hours (20 Primary Manufacturers × 1,000 hr/response × 1 response/year) at a cost of $2,281,704.00 (20 responses × [(200 hr x $95.06/hr + (600 hr × $119.92/hr) + (50 hr × $175.98/hr) + (4 hr × $285.14/hr) + (142 hr × $82.82/hr) + (4 hr × $355.30/hr)].
Table 9—Summary of Total Annual Burden for Completion of Negotiation Data Elements ICR Form for Selected Drugs for Negotiation by Primary Manufacturers
Requirement
Number of
respondents
Total annual responses
Time per
response
(hours)
Total annual time
(hours)
Labor
cost
($/hr)
Total cost
($)
Negotiation Data Elements for Selected Drugs for Negotiation from Primary Manufacturers
20
20
1,000
20,000
Varies
2,281,704.00
( printed page 36327)
(2) Negotiation Data Elements for Selected Drugs for Renegotiation From Primary Manufacturers
In accordance with section 1194(f)(4)(B) of the Act and as proposed at § 429.615(b) of this proposed rule, along with definitions proposed in § 429.20, CMS will apply a similar approach regarding data collection once a drug is selected for renegotiation of the MFP, if any drugs are selected for renegotiation. Although information submission for factors outlined in section 1194(e)(2) of the Act are voluntary and open to all interested parties, this burden estimate assumes that all Primary Manufacturers will choose to submit this type of data. CMS assumes a lower burden for data submissions for drugs selected for renegotiation relative to the original submission of data for negotiation because Primary Manufacturers will need to report only new data for a short period of time.
CMS estimates collecting up to 36 responses from Primary Manufacturers of selected drugs for burden estimate base year initial price applicability year 2029 for renegotiation. CMS anticipates that fewer than 36 drugs will be selected for renegotiation, but CMS cannot provide definitive assumptions about how many drugs may be selected for renegotiation. The collection of these data will be a one-time cost for each selected drug and CMS assumes each Primary Manufacturer will spend, on average, the same amount of time to collect, aggregate, analyze, and report the data for a selected drug.
Using the wage rates in Table 5 of this proposed rule, we expect, for a Primary Manufacturer, a business operations specialist or team of business operations specialists 150 hours to gather cost data and compile required information, as specified in the data elements instructions, an economist or team of economists 450 hours to perform necessary economic analyses of data elements specified in the data element instructions, a financial manager 37.5 hours to review the results of all the analyses and cost estimates prior to submission to CMS, a lawyer 3 hours to review the compiled data submission, a cost estimator 106.5 hours to compile and report the required data to CMS, per the data element form instructions, and a chief executive 3 hours to review the data submission and log in to the CMS HPMS to certify the submission.
In aggregate, we estimated an annual burden of 27,000 hours (36 Primary Manufacturers × 750 hr/response × 1 response/year) at a cost of $3,080,300.40 (36 responses × [(150.0 hr × $95.06/hr + (450.0 hr × $119.92/hr) + (37.5 hr × $175.98/hr) + (3.0 hr × $285.14/hr) + (106.5 hr × $82.82/hr) + (3.0 hr × $355.30/hr)].
Table 10—Summary of Total Annual Burden Completion of Negotiation Data Elements ICR Form for Selected Drugs for Renegotiation by Primary Manufacturers
Requirement
Number of
respondents
Total annual
responses
Time per
response
(hours)
Total annual time
(hours)
Labor
cost
($/hr)
Total cost
($)
Selected Drugs for Renegotiation from Primary Manufacturers
36
36
750.0
27,000
Varies
3,080,300.40
(3) Negotiation Data Elements for Selected Drugs for Negotiation and Renegotiation from the General Public
To generate burden estimates for the section 1194(e)(2) data collection for burden estimate base year initial price applicability year 2029, CMS reviewed the public feedback that was received for the initial price applicability year 2028 negotiation period and adjusted accordingly due to the inclusion of selected drugs for renegotiation and the year-over-year greater awareness around the public input process for the Drug Price Negotiation Program.
CMS estimates collecting a total of 325 requests from the general public for initial price applicability year 2029. We believe that approximately 150 individual respondents and 175 organizations will respond because the proportion of individuals and organizations remained generally consistent across the first two initial price applicability years for which data has been collected thus far.
Using the wage rates in Table 5 of this proposed rule, we expect, for an individual respondent to spend 3 hours to review literature and submit information to CMS for a selected drug and for an organization to spend 30 hours to review literature and submit information to CMS for a selected drug.
In aggregate, we estimated an annual burden of 5,700 hours [(150 individuals × 3 hr/response × 1 response/year) + (175 organizations × 30 hr/response × 1 response/year)] at a cost of $279,414.00 [(150 responses × 3 hr × $49.02/hr) + (175 responses × 30 hr × $49.02/hr)].
Table 11—Summary of Total Annual Burden for Completion of Negotiation Data Elements ICR Form for Selected Drugs for Negotiation and Renegotiation by the General Public
Requirement
Number of
respondents
Total annual
responses
Time per response
(hours)
Total annual time (hours)
Labor cost
($/hr)
Total cost
($)
Selected Drugs for Negotiation and Renegotiation from the General Public
325
325
Varies
5,700
49.02
279,414.00
(4) Temporary Floor for Small Biotech Drugs
This ICR also collects information necessary for CMS to determine eligibility of a selected drug, for negotiation or renegotiation in initial price applicability years 2029 or 2030, for the temporary floor of the maximum fair price provided to selected drugs determined by CMS to be small biotech drugs consistent with section 1194(d) of the Act and as proposed at § 429.445 (the “Temporary Floor for Small Biotech Drugs”) of this proposed rule. Because eligibility for the Temporary Floor for Small Biotech Drugs relies on the same eligibility criteria specified in section 1192(d)(2) of the Act to determine if an otherwise negotiation-eligible drug was excluded from selection for initial price applicability years 2026, 2027, or 2028 because the drug met the qualifications for the exception for small biotech drugs
( printed page 36328)
(the “small biotech exception” or “SBE”), CMS is repurposing the SBE ICR form (CMS-10844) to now collect the applicable information for the Temporary Floor for Small Biotech Drugs in this ICR (CMS-10849) for initial price applicability years 2029 and 2030.
CMS estimates up to 10 respondents will need to submit data to determine applicability of the Temporary Floor for Small Biotech Drugs per year. We believe that the collection of these data will be a one-time cost for a Primary Manufacturer for each selected drug that may be eligible for the Temporary Floor for Small Biotech Drugs for initial price applicability years 2029 or 2030.
Using the wage rates in Table 5 of this proposed rule, we expect, for a Primary Manufacturer, a lawyer 6.50 hours to gather and review the relevant ICR provisions, identify any controlled group members, and request technical assistance from CMS, a general and operations manager 3.00 hours to examine the gathered information and submit the ICR Form for eligibility for the Temporary Floor for Small Biotech Drugs to CMS and request technical assistance from CMS, and a chief executive 0.25 hour to review the information prior to submission and to log into CMS' existing information technology system to certify the submission.
In aggregate, we estimated an annual burden of 97.5 hours (10 Primary Manufacturers × 9.75 hr/response × 1 response/year) at a cost of $24,401.75 (10 responses × [(6.50 hr × $285.14/hr + (3.00 hr × 165.98/hr) + (0.25 hr × $355.30/hr)].
Table 12—Summary of Total Annual Burden for Completion of Temporary Floor for Small Biotech Drugs ICR Form
Requirement
Number of
respondents
Total annual
responses
Time per
response
(hours)
Total annual time
(hours)
Labor cost
($/hr)
Total cost
($)
Temporary Floor for Small Biotech Drugs
10
10
9.75
97.5
Varies
24,401.75
(5) Counteroffer
A Primary Manufacturer must complete and submit the information requested on the Statutory Written Counteroffer ICR Form or the Renegotiation Written Counteroffer ICR Form, as applicable, if it both chooses not to accept CMS' initial offer and chooses to submit a Counteroffer for a selected drug. The Statutory Written Counteroffer ICR Form and the Renegotiation Written Counteroffer ICR Form are collectively referred to as the “Counteroffer ICR Form” due to CMS approximating the estimates for each form to be similar due to the questions on the forms requiring about the same amount of time for a manufacturer to collect and submit the information on the applicable form.
CMS estimates collecting up to 56 responses from Primary Manufacturers for burden estimate base year initial price applicability year 2029. CMS chose this number because by statute only up to 20 drugs payable under Medicare Part B and/or covered under Medicare Part D can be selected for negotiation for 2029 and a maximum of 36 drugs can be selected for renegotiation for initial price applicability year 2029, and for each selected drug CMS will undergo negotiation or renegotiation with only one Primary Manufacturer, so it is not possible that there would be more than 57 respondents for initial price applicability year 2029.
Using the wage rates in Table 5 of this proposed rule, we expect, for a Primary Manufacturer, a business operations specialist or team of business operations specialists 27.00 hours to review CMS' initial offer and justification and compare it to current prices, revenue, and other market and clinical data for the selected drug and compare CMS' justification with the data the Primary Manufacturer submitted as part of the section 1194(e)(1) and 1194(e)(2) factors and the section 1194(e)(2) data from other interested parties shared by CMS with the Primary Manufacturer, if feasible, and put together recommendations on how the initial offer compares to what was submitted and develop Counteroffer options and justifications, a team of healthcare professionals (a pharmacist 15.00 hours, a registered nurse 5.00 hours, and a general internal medicine physician 5.00 hours) 25.00 hours to compare CMS' initial offer and justification to the section 1194(e)(2) factors around the selected drug and therapeutic alternatives and develop Counteroffer options and justifications, an economist or team of economists 64.00 hours to consider team recommendations of the business operations specialist(s) and healthcare professionals, model counteroffer options, and recommend Counteroffer options, a general and operations manager or team of general and operation managers 14.25 hours to review Counteroffer options and justifications and develop a Counteroffer proposal for the MFP and to examine the gathered information, populate the Counteroffer ICR Form, and submit the Counteroffer ICR Form to CMS, a lawyer or team of lawyers 64.00 hours to review counteroffer options and draft a justification for the selected Counteroffer proposal for the MFP, and a chief executive 10.00 hours to review the Counteroffer proposal for the MFP, make a decision on the Counteroffer proposal for the MFP, review the Counteroffer information prior to submission, and log in to the CMS HPMS to certify the submission.
In aggregate, we estimated an annual burden of 11,438 hours (56 Primary Manufacturers × 204.25 hr/response × 1 response/year) at a cost of $2,127,987.40 (56 responses × [(27.00 hr × $95.06/hr + (15.00 hr × $133.46/hr) + (5.00 hr × $71.14/hr) + (5.00 hr × $246.70/hr) + (64.00 hr × $119.92/hr) + (14.25 hr × $165.98/hr) + (64.00 hr × $285.14/hr) + (10.00 hr × $355.30/hr)].
Table 13—Summary of Total Annual Burden for Completion of Counteroffer ICR Form
Requirement
Number of
respondents
Total annual
responses
Time per
response
(hours)
Total annual time
(hours)
Labor cost
($/hr)
Total cost
($)
Counteroffer
56
56
204.25
11,438
Varies
2,127,987.40
( printed page 36329)
Table 14—Summary of Total Annual Burden for Drug Price Negotiation ICR
Drug Price Negotiation for Initial Price Applicability Year 20XX under Sections 11001 and 11002 of the Inflation Reduction Act (using base year initial price applicability year 2029)
0938-1452 (CMS-10849)
447
447
Varies
64,235.50
Varies
7,793,807.55
C. Summary of Annual Burden Estimates
Table 15 sets out the burden for this rulemaking's finalized provisions that are subject to the PRA. It does not score burden adjustments that are strictly based on updated data and are unrelated to any of the provisions.
Table 15—Annual Requirements and Burden Estimates
[With Base Year Initial Price Applicability Year 2029]
§§ 429.110, 429.610, and 429.615 Negotiation Program Drug Selection for Initial Price Applicability Year 20XX under Sections 11001 and 11002 of the Inflation Reduction Act
0938-1443 (CMS-10844)
46
46
Varies
4,760
Varies
582,859.20
§§ 429.110, 429.610 and 429.615 Drug Price Negotiation for Initial Price Applicability Year 20XX under Sections 11001 and 11002 of the Inflation Reduction Act
0938-1452 (CMS-10849)
447
447
Varies
64,235.50
Varies
7,793,807.55
Total
493
493
Varies
68,995.50
Varies
8,376,666.75
V. Regulatory Impact Analysis
A. Statement of Need
This proposed rule proposes to codify, with limited modification, policies related to the implementation of certain provisions of the Inflation Reduction Act of 2022 (IRA) (Pub. L. 117-169, August 16, 2022), which were established in the final guidance for the Medicare Drug Price Negotiation Program (hereinafter the “Negotiation Program”). The primary driver for this rulemaking is the statutory mandate to codify changes made by the IRA. Without regulatory implementation of the Negotiation Program, the Medicare program cannot comply with Federal law or reduce prescription drug costs for beneficiaries or achieve cost savings provided under statute.
This rulemaking proposes to codify policies for the Negotiation Program at part 429 consistent with sections 1191 through 1198 of the Social Security Act (hereinafter “the Act”) and to codify policies for the Medicare Prescription Drug Benefit Program at part 423 consistent with section 11001(b) of the IRA, which made certain amendments to the Act, including with respect to Medicare Part D. Our policies in this rulemaking specifically address: general provisions; identification of selected drugs; Negotiation Program Agreement; program administration; establishment of a single maximum fair price (MFP) and determination of the ceiling; negotiation process; renegotiation of an MFP; implementation of the MFP; manufacturer compliance and oversight; civil monetary penalties; and requirements related to qualified prescription drug coverage. In addition, we are proposing new policies for the Negotiation Program as follows: a narrow modification to the general fixed combination drug policy to clarify our treatment for certain fixed combination drugs that are new formulations; clarification related to how CMS would identify the day from which to measure the 7- and 11-year time since approval and licensure periods for drugs that formerly qualified for the orphan drug exclusion; revisions regarding the process and schedule of CMS review of information in making determinations with respect to Bona Fide Marketing; additional details related to the Primary Manufacturer transfer of responsibility for all requirements of the Negotiation Program Agreement to an acquiring entity; calculation of the 30-day equivalent supply for a selected drug that is typically administered one time; how CMS would implement the Temporary Floor for Small Biotech Drugs for initial price applicability years 2029 and 2030; and clarification of off-label use in consideration for renegotiation eligibility and selection. The policies reflect CMS' stewardship of the Medicare program and overarching policy objectives for lower prices for Medicare through negotiation or price competition by lowering drug prices and making Medicare prescription drugs more affordable for beneficiaries.
The absence of regulatory action would result in statutory non-compliance regarding IRA implementation and missed opportunities for program improvement and innovation. Therefore, this rulemaking is necessary to ensure the Negotiation Program operates effectively, efficiently, and in compliance with Federal law while serving the best interests of Medicare beneficiaries.
B. Overall Impact
We have examined the impacts of this rule as required by Executive Order 12866, “Regulatory Planning and Review”; Executive Order 13132, “Federalism”; Executive Order 13563, “Improving Regulation and Regulatory Review”; Executive Order 14192, “Unleashing Prosperity Through Deregulation”; the Regulatory Flexibility Act (RFA) (Pub. L. 96 354); section 1102(b) of the Social Security Act; and section 202 of the Unfunded
( printed page 36330)
Mandates Reform Act of 1995 (Pub. L. 104-4).
Executive Orders 12866 and 13563 direct agencies to assess all costs and benefits of available regulatory alternatives and, if regulation is necessary, to select those regulatory approaches that maximize net benefits (including potential economic, environmental, public health and safety, and other advantages; distributive impacts.). Section 3(f) of Executive Order 12866 defines a “significant regulatory action” as any regulatory action that is likely to result in a rule that may: (1) have an annual effect on the economy of $100 million or more or adversely affect in a material way the economy, a sector of the economy, productivity, competition, jobs, the environment, public health or safety, or State, local, or tribal governments or communities; (2) create a serious inconsistency or otherwise interfere with an action taken or planned by another agency; (3) materially alter the budgetary impact of entitlements, grants, user fees, or loan programs or the rights and obligations of recipients thereof; or (4) raise novel legal or policy issues arising out of legal mandates, or the President's priorities.
A regulatory impact analysis (RIA) must be prepared for a regulatory action that is significant under section 3(f)(1) of E.O. 12866. Based on our estimates, OIRA has determined that this rulemaking is “significant” under section 3(f)(1) of E.O. 12866.
We have prepared an RIA that, to the best of our ability, presents the costs and benefits of the rulemaking.
C. Detailed Economic Analysis
The CMS Office of the Actuary estimated the impacts of the drug provisions of the IRA using the 2024 President's Budget as a baseline early in calendar year 2023. These estimates were made prior to many policy decisions to implement the law, and independently from other components of CMS. Since the majority of these provisions have already been implemented through program instruction, including Medicare Drug Price Negotiation Program guidance with respect to initial price applicability years 2026, 2027, and 2028 (as discussed in further detail in section I.A.2. of this proposed rule), this estimate should be taken only in its historical context, not as a reflection on the experience since its effectuation. We will highlight certain components of this estimate where recent experience has diverged from our initial assumptions. The IRA has a range of Medicare provisions, including restraining price growth and negotiating drug prices for certain drugs payable under Part B and covered under Part D, as well as redesigning the Part D benefit structure to decrease beneficiary out-of-pocket costs. The provisions of the IRA take effect over several years, resulting in very different effects by year. Much of the Part D benefit redesign became effective in 2025, for example, before the Negotiation Program provisions can have any offsetting effects.
To model the negotiation provisions of the IRA, we first determined which drugs would be selected for negotiation in accordance with sections 11001 and 11002 of the IRA. Using 2022 experience for Part B and Part D claims, we ranked drugs by Part B and Part D expenditures and then applied the eligibility criteria specified in the IRA—verifying, in particular, that the ranked drugs had been on the market long enough to qualify for negotiation. From this list, we generated the potential list of drugs to be negotiated in each year for Part B and Part D.
To estimate the impact of negotiation and to measure the differences between the current prices and the ceiling price and other pricing parameters laid out in the IRA, we used 2021 data from a variety of sources, including PDE records, Medicaid AMP data, and Part B ASP data. We assumed, after comparing the Medicare prices to the ceiling prices in each projection year, that Medicare would be able to negotiate slightly below the ceiling price in Part D. We then adjusted for generic and biosimilar launches that, should they happen after the selection process, would potentially limit the impact of the maximum fair price. Lastly, we adjusted for changes in the percentage of spending that the selected drugs would represent over time. The discounts relative to total 2021 Part D allowed cost, prior to manufacturer rebates and total Part B allowed cost, are shown in Table 16.
Table 16—Cumulative Negotiation Discount From Allowed Costs
2026
%
2027
%
2028
%
2029
%
2030
%
2031
%
Part B
0
−1
−5
−6
−9
−12
Part D
−9
−16
−20
−23
−24
−25
For Part D, the benefit is considerably enriched under the IRA, and the most impactful changes took effect in 2025. To estimate these effects inclusive of the Negotiation Program impacts, we incorporated the negotiated price at the drug level into a beneficiary- and claim-level detailed model. Then, we recalculated the new benefit on the negotiated prices to determine the combined result under the defined standard benefit design by year. To protect beneficiaries from large premium increases, the IRA limits the premium change in years 2024 through 2029 before ultimately readjusting the base beneficiary premium percentage to a minimum of 20 percent in 2030 and later years. The major benefit changes and beneficiary premium protections by year are shown in Table 17.
Table 17—IRA Part D Benefit and Premium Changes
2023
2024
2025-2029
2030
Benefits
Reduced cost sharing for insulins and Advisory Committee on Immunization Practices (ACIP)-recommended adult vaccines
No cost sharing in catastrophic phase
Removal of coverage gap, $2,000 out-of-pocket max (indexed), new manufacturer discount
No benefit changes.
Premium
Funded by retrospective government subsidy
Base beneficiary premium (BBP) limited to 6% increase from prior year
BBP limited to 6% increase from prior year
BBP equal to lesser of prior year's BBP increased by 6% or the BBP calculated under section 1860D-13(a)(2) of the Act, with BBP not less than 20% of bid and reinsurance costs.
( printed page 36331)
To complete the modeling of the Negotiation Program, prescription drug inflation rebates, and benefit provisions, we applied the results from the Part D claim-level detailed simulation to our total Part D benefit model. This model also incorporated changes to the per capita cost trends to reflect: (i) the impact of most existing brand-name drugs moving to a CPI-level increase; (ii) the expected growth in new drug costs; and (iii) the expected induced utilization due to the enriched benefit. We assumed that drugs with a significant amount of spending in Part D would temper their price increases rather than pay the inflation rebates required by the IRA. Additionally, we reduced our estimate of manufacturer rebates to compensate for the lower negotiated prices and lower price growth on existing drugs.
For drugs payable under Part B, we applied the negotiated price discounts to the OM spending for separately payable Part B drugs. We further adjusted these results to account for the impact to private health plan expenditures to obtain a total impact. While there are also inflation rebates required for certain drugs payable under Part B under the IRA, price increases on existing drugs payable under Part B historically have been close to the CPI in aggregate, and therefore we did not project an effect for this provision in the drugs payable under Part B. We also incorporated other, less significant changes from the IRA, such as the lower cost sharing for insulins furnished under durable medical equipment and the temporary payment increase for qualifying biosimilar products.
Our assumptions on the impacts of the IRA differed from those used in other public estimates in a few critical ways. Most importantly, we assumed that the inflation rebates required by the IRA would result in manufacturers owing relatively small inflation rebate amounts for drugs covered under Part D and nothing for drugs payable under Part B. We assumed that manufacturers would prefer to have lower price trends that would incentivize greater use than pay publicly reported fees for price increases that exceed inflation. Under this assumption, the effects of the inflation rebate provisions of the IRA are changed because the difference in price is shared across the benefit rather than accruing directly to the government. In other words, lower list price trends will reduce prices paid at the pharmacy relative to the baseline, which results in lower beneficiary cost-sharing and lower state clawback payments, thereby increasing the Federal cost for the Part D benefit. To compensate for the loss of price increases, we expected manufacturers to reduce rebates offered to plan sponsors.
Additionally, we expected that the initial pool of drugs covered under Part D selected for negotiation would have a large proportion of heavily rebated drugs. In these cases, we expected that the price net of rebate will be substantially lower than the other ceiling prices described in the IRA. We further estimated that the effect of negotiation in the early years would be similar to the impact of shifting rebates to the point of sale. This shift reduces beneficiary cost sharing as the price at the point of sale is lower but increases bid amounts and increases Federal expenditures.
In summary, the total effects were to reduce government expenditures for Part B, to increase expenditures for Part D through 2030, and to decrease Part D expenditures beginning in 2031. Part B savings were primarily due to: (i) the substantial lowering of payments, relative to current payment, as a result of maximum fair prices; and (ii) small impacts from other provisions. Part D ultimately generated cost savings at the end of the budget window, but many of the gains from maximum fair prices and lower trends were initially spent on increased benefits and the loss of manufacturer rebates. The impact on benefits and premiums and the impact in total are shown in Table 18 using the 2024 President's Budget as a basis.
Table 18—Part B and Part D IRA Impacts
[In billions]
2023
2024
2025
2026
2027
2028
2029
2030
2031
2032
Part B:
Total benefits
$0.1
$0.1
$0.1
−$0.2
−$0.3
−$5.8
−$8.4
−$14.7
−$21.4
−$28.4
Premium
0.0
0.0
0.0
−0.1
−0.1
−1.5
−2.1
−3.7
−5.3
−7.1
Total Federal impact
0.1
0.1
0.1
−0.2
−0.2
−4.4
−6.3
−11.0
−16.1
−21.3
Part D:
Total benefits
0.9
6.5
9.5
11.3
7.5
3.0
0.4
−4.5
−10.4
−12.5
Premium
0.0
−0.3
−0.6
−0.3
0.2
0.7
1.3
0.7
−0.1
−0.3
Inflation rebates
0.4
0.3
0.3
0.2
0.2
0.2
0.2
0.3
0.3
0.3
State transfer impact
0.0
−0.1
−0.4
−0.6
−1.7
−3.5
−5.1
−6.4
−7.6
−8.6
Total Federal impact 1
0.4
6.6
10.2
11.9
8.8
5.6
3.9
1.0
−3.0
−3.9
The Federal impact is calculated as the benefit impact less: (i) the premium impact; (ii) the inflation rebate impact; and (iii) the State transfer impact.
Note:
Totals do not necessarily equal the sums of rounded components.
Since we produced these estimates, both the Part B and Part D programs have had large increases in drug expenditures. Part D trends have accelerated dramatically, with the per capita gross drug cost increasing more than 18 percent in 2025 over 2024, driven by higher glucagon-like peptide-1 (GLP-1) and specialty drug usage. Meanwhile Part B drug trends have also increased, although the primary driver was skin substitutes that were payable as biologicals for the purposes of Medicare payment. There are a variety of other causes contributing to observed changes in expenditures for both programs.
The Part D trends in per capita gross costs are shown in Figure 3. Much of the IRA Part D benefit redesign took effect in 2025, including limiting annual out of pocket expenditures to $2,000 per enrollee for 2025 (to be annually increased by the annual percentage increase, as described in section 1860D-2(b)(6) of the Act). While this new feature could have induced spending beyond the assumption we included in our initial estimate, it is worth noting that the 2024 benefit structure under the IRA also eliminated cost-sharing in the catastrophic phase of the benefit, effectively implementing an out-of-pocket maximum without a pronounced increase in costs. Additionally, some of the increase may be attributable to manufacturers reducing spending on patient assistance programs, which would cause more claims to run through the Part D program. Expanded indications for cancer drugs also contributed to the increase.
Figure 3: Part D per Capita Gross Drug Costs
( printed page 36332)
The observed discounts from MFPs for selected drugs also differed from what we initially assumed. For initial price applicability year 2026, the projected negotiated discount impact on allowed costs—ingredient cost, dispensing fee, and sales tax—using 2025 experience data is 11.3 percent, while the projected impact for initial price applicability year 2027 is 18.2 percent. Compared with the original estimated effects shown in Figure 3, the actual negotiated discounts are more than 2 percent greater than the original modeling results. These differences reflect the deviation in price levels, updated information about generic and biosimilar launches, and the percentage of expense the negotiated drugs represent for each year. Part B MFPs for initial price applicability year 2028 are not currently available, so we do not quantify the change from our original assumptions. We also had assumed that MFPs for initial price applicability years 2026 and 2027 would also apply to Part B utilization. Final policies for those years contained the MFPs to the Part D benefit. This change from our assumption did not meaningfully affect overall estimates, due to multiple factors including limited observed utilization of most initial price applicability years 2026 and 2027 drugs in Part B and the timing of certain generic or biosimilar entrants. Until the entirety of 2026 Part D data is available, including Part D Direct and Indirect Remuneration data, we are unable to determine the ultimate impact of initial price applicability year 2026 MFPs. Even after such data is available, there will not be an accurate way to attribute elements of the experience to particular provisions. For example, the 2026 claims experience will undoubtedly be impacted by the benefit redesign changes implemented in 2025, but determining what portion of 2026 results are due to those benefit changes compared with the implementation of MFPs is unclear.
Part D drug inflation rebates were higher than originally assumed. Converting the published inflation rebates owed from the applicable period to a calendar year basis, 2023 inflation rebates were over $500 million dollars. This is higher than the $400 million dollar estimate shown in Table 18. This implies that drug prices increased slightly faster than originally assumed, generating higher inflation rebates. As a percentage of gross drug costs, the original estimate represents 0.01 percent, while the actuals amount to 0.02 percent.
For Part B, our original estimates assumed that the drug inflation rebates would be negligible. Actual drug inflation rebates due for calendar years 2023 and 2024 were $14 million and $121 million, respectively. These amounts represent approximately 0.01 percent and 0.04 percent of the incurred charges for OM in 2023 and 2024. Since drug inflation rebates are not incurred on MA utilization at this time, this is the most appropriate comparison.
Other, more nuanced elements of the IRA changes also differed from our original expectations. For example, the IRA Manufacturer Discount Program is likely larger than we initially assumed, but the PDE reporting is not yet complete for the discount's first year. Similarly, the impacts of the IRA on Part D DIR are unknown, as Part D plan sponsors have not yet submitted the 2025 DIR reports. These elements may have larger effects on overall Part D expense than the other assumption deviations previously described.
Additionally, this rule implements provisions of the “Working Families Tax Cuts Act” (“WFTC Act”) (Pub. L. 119-21) of 2025. This legislation expanded the IRA's orphan drug exclusion, allowing drugs that are designated as a drug for one or more rare diseases or conditions under section 526 of the FD&C Act and approved by FDA only for indications within such designated rare disease(s) or condition(s) to be excluded from the Negotiation Program effective with respect to initial price applicability year 2028. Additionally, and also effective with respect to initial price applicability year 2028, the WFTC Act amended how CMS identifies the day from which to measure the 7- and 11-year time since approval and licensure periods for drugs that formerly qualified for the orphan drug exclusion. These amendments lead to changes in projections of which drugs will be eligible for negotiation and corresponding changes to the overall effects of negotiation.
We analyzed these effects using baseline estimates from the 2026 Mid-Session Review budget exercise. Following the general procedures described previously for estimating the effects of the IRA negotiation provisions, we adjusted the projected drugs to be negotiated by year to account for the WFTC Act amendments. Orphan drug exclusions were determined using data from the FDA on orphan drug designations. We did not forecast the likelihood of these drugs losing their orphan status, as we assume
( printed page 36333)
the relative value of orphan and non-orphan drugs remains stable over time. While it is possible that some drugs will avoid non-orphan indication approvals to take advantage of this exclusion, these situations will vary widely from drug to drug, and we do not have a basis to assume the future effects of such behavior.
Table 19 shows the impacts to Medicare Part B and Part D benefits and premiums. These impacts are reflected in accounting Table 19.
Table 19—Part B and Part D WFTC Act Impacts
[in billions]
2028
2029
2030
2031
2032
2033
2034
Part B:
Total benefits
$2.8
$2.7
$0.2
$0.5
$0.4
$1.3
$1.6
Premium
0.7
0.7
0.1
0.1
0.1
0.3
0.4
Total Federal impact
2.1
2.0
0.2
0.3
0.3
0.9
1.2
Part D:
Total benefits
−0.2
0.0
0.1
0.0
0.4
0.8
1.0
Premium
0.0
0.0
0.0
0.0
0.1
0.1
0.2
Inflation rebates
0.0
0.0
0.0
0.0
0.0
0.0
0.0
State transfer impact
0.0
0.0
0.0
0.0
0.0
0.0
0.0
Total Federal impact 1
−0.2
0.0
0.1
0.1
0.4
0.6
0.9
1. Analysis of New Policies
In addition, we are proposing new policies for the Negotiation Program as follows: a narrow modification to the general fixed combination drug policy to clarify our treatment of certain new formulations; clarification related to how CMS would identify the day from which to measure the 7- and 11-year time since approval and licensure periods for drugs that formerly qualified for the orphan drug exclusion; revisions regarding process and schedule of CMS review of information in making determinations for Bona Fide Marketing; additional details related to the Primary Manufacturer transfer of responsibility for all requirements of the Negotiation Program Agreement to an acquiring entity; calculation of the 30-day equivalent supply for a selected drug that is typically administered one time; how CMS would implement the Temporary Floor for Small Biotech Drugs for initial price applicability years 2029 and 2030; and clarification of off-label use in consideration for renegotiation eligibility and selection. Of these proposals, only the proposed narrow modification to the general fixed combination drug policy would potentially have an impact as the other provisions are technical changes that would not result in additional costs or savings to the Negotiation Program.
The fixed combination drug proposal involves costs and savings that extend outside the budget window for this regulatory impact analysis. For purposes of this discussion, we refer to a potential qualifying single source drug containing active moiety/active ingredient/antigen component X as Product A, and a new formulation of such potential qualifying single source drug containing active moiety/active ingredient/antigen component X plus active moiety/active ingredient/antigen component Y, which meets the criteria proposed at § 429.125(b)(4)(i), as Product B. While a narrow modification to the fixed combination drug policy that aggregates Product A and Product B into the same qualifying single source drug would, if such qualifying single source drug were selected, result in Product B being part of the selected drug and thus having any agreed upon MFP applied earlier, this narrowly modified fixed combination policy would also expand the number of products aggregated under a selected drug that would become deselected once a generic or biosimilar is subject to Bona Fide Marketing for any dosage form or strength of the selected drug, resulting in Product B potentially having the agreed-upon MFP applied for a shorter period of time than if it had been identified as a separate qualifying single source drug that was selected and negotiated separately. Therefore, potential savings generated by aggregating more products into a qualifying single source drug to which, if selected, the agreed-upon MFP would apply could be limited due to corresponding potential savings lost by the larger number of products that would be deselected once a generic or biosimilar is subject to Bona Fide Marketing, though changes in price after the drug is deselected may be contained (and thus savings preserved) by the Inflation Rebate Program for drugs payable under Part B.[81]
In addition, a narrow modification to the fixed combination drug policy that would aggregate Product A and Product B into the same potential qualifying single source drug could result in a scenario where Product A has a generic or biosimilar that is subject to Bona Fide Marketing before that drug's eligibility for selection, which could result in both Product A and Product B being ineligible for selection for negotiation, meaning that an MFP could not be negotiated for either product.
As an illustrative example, if a drug selected for negotiation for initial price applicability year 2029 has a new formulation that is a fixed combination drug containing hyaluronidase, the proposed policy would include the new formulation containing hyaluronidase in the selected drug if the criteria in proposed § 429.125(b)(4)(i) were met. In this case, the MFP, if one is agreed to by CMS and the Primary Manufacturer, would apply to all dosage forms and strengths of the qualifying single source drug, including the new formulation containing hyaluronidase, in initial price applicability year 2029 and each subsequent year unless and until CMS makes a determination in accordance with § 429.135(a) that such drug ceases to be a selected drug because a manufacturer for a biosimilar for the non-hyaluronidase product engages in Bona Fide Marketing. Alternatively, if we do not finalize the proposal, the new formulation containing hyaluronidase might be separately selected for negotiation in 2039 (an illustrative year to represent the earliest date when the 11-year timing criterion set forth in proposed § 429.125(c)(2) for a biological product has been reached). These effects would be outside the budget window, and we do not have a credible, drug-
( printed page 36334)
level forecast for that time period. Such a forecast would be required to estimate this provision, as the impacted set of drugs is small and the effects will depend on the level of utilization of this subset of drugs far in the future. Additionally, we are unable to accurately estimate the impact of finalizing the narrow modification to the general fixed combination drug policy within the budget window since the budgetary effects of the counterfactual (that is, not finalizing such proposal) are outside the budget window, and we do not have baseline estimates for that time horizon. We welcome comments on methodologies to estimate these effects.
D. Alternatives Considered
In this section, CMS includes discussions of alternatives considered.
1. Fixed Combination Products (§ 429.105(b)(4))
The first alternative we considered would further narrow the proposed modification to the application of the Negotiation Program's general fixed combination drug policy to biological products only. As compared to the policy proposed at § 429.125(b)(4), this alternative may not meaningfully impact which products would be aggregated under a qualifying single source drug.
The second alternative we considered would modify the general fixed combination drug policy for fixed combination drugs for which one of the active ingredients or active moieties contained is not biologically active against the disease state(s) the drug is indicated for and thus does not result in a clinically meaningful difference. As compared to the policy proposed at § 429.125(b)(4), this alternative may not meaningfully impact which products would be aggregated under a qualifying single source drug.
The third alternative we considered would maintain the policy established for initial price applicability years 2026 through 2028, wherein, for a fixed combination drug with two or more active moieties or active ingredients, we would treat the distinct combination of active moieties or active ingredients as one active moiety or active ingredient. As compared to the policy proposed at § 429.125(b)(4), this alternative may reduce the number of products that would be aggregated under a qualifying single source drug. If we maintained the policy established for initial price applicability years 2026 through 2028, selection and negotiation for a new formulation of a qualifying single source drug could be delayed, which might reduce total savings under the Negotiation Program.
Because these alternatives considered involve financial impacts outside the 10-year budget window and we do not have credible drug-level forecast that far out, quantifying longer-term effects would be speculative, and only considering impacts within the window would not adequately explain the full results of these alternatives.
Further discussion of this proposal is included in section II.B.6. of this proposed rule.
E. Accounting Statement and Table
Consistent with OMB Circular A-4 (available at
https://www.reginfo.gov/public/jsp/Utilities/a-4.pdf), we have prepared an accounting statement in Table 20 showing the classification of the impact associated with the provisions of this rule.
Table 20—Accounting Statement
[$ millions]
Category
3% Discount
rate
7% Discount
rate
Notes
Transfers:
From Federal Government and Medicare Enrollees to pharmaceutical manufacturers
9,976
8,695
Orphan drug provision.
F. Regulatory Flexibility Act (RFA)
The Regulatory Flexibility Act (RFA) requires agencies to analyze options for regulatory relief for small entities. For purposes of the RFA, small entities include small businesses, nonprofit organizations, and small governmental jurisdictions. Individuals and states are not included in the definition of a small entity. The RFA requires that CMS analyze regulatory options for small businesses and other entities unless CMS certifies that a rule would not have a significant economic impact on a substantial number of small entities. The analysis must include a justification concerning the reason action is being taken, the kinds and number of small entities the proposed rule effects, and an explanation of any meaningful options that achieve the objectives with less significant adverse economic impact on the small entities.
HHS considers a significant impact on a substantial number of small entities to be one with a 3 percent revenue effect on 5 percent of small entities.[82]
CMS would consider a “significant impact” to be 3 percent or more of the affected entities' costs or revenues, and a “substantial number” to mean 5 percent or more of affected small entities within the total number of the small businesses in that North American Industry Classification System (NAICS) industry description. As discussed in section II of this proposed rule, the Medicare Drug Price Negotiation Program requires the Secretary to negotiate and renegotiate, for applicable periods, Medicare prices for certain high expenditure, single source drugs and biological products. Our analysis shows that the proposed rule may directly impact one category of small entities, Primary Manufacturers. Given the uncertainty on negotiation outcomes and available data, CMS concludes that this proposed rule, if finalized as proposed, may have an economic impact on small entities. This analysis as well as other sections in this proposed rule, serves as the Initial Regulatory Flexibility Analysis, as required by the RFA.
1. Description and Number of Affected Small Entities
We use the NAICS to identify the industry potentially affected by the proposed rule. We also use the Small Business Administration (SBA) size standards in “number of employees” and “millions of dollars” to identify small entities.[83]
The SBA considers any “Pharmaceutical Preparation Manufacturing” firm (NAICS code 325412) with fewer than 1,300 employees as a small business. The most recent data on private sector
( printed page 36335)
entities with an NAICS code 325412 at the time this proposed rule was drafted are from the 2022 Statistics of U.S. Businesses (SUSB),[84]
which reports 1,179 private firms and 1,444 private establishments with paid employees. Of these, 1,049 firms and 1,100 establishments have fewer than 500 employees, while 130 firms and 344 establishments have more than 500 employees as shown in Table 21. The SUSB data does not provide an enterprise range that presents the number of firms or establishments with fewer than 1,300 employees, however the numbers presented show that 82 percent of the combined firms and establishments have less than 500 employees while 18 percent have 500 or more employees.
Table 21—SUSB Firms, Establishments, Employment Size, and Receipts Data
Enterprise size
Firms
Establishments
Employment
Receipts
($1,000)
<5 employees
388
388
678
782,552
5-9 employees
143
145
972
425,072
10-19 employees
126
127
1,638
983,091
<20 employees
657
660
3,288
2,190,715
20-99 employees
219
225
9,633
4,276,317
100-499 employees
173
215
31,673
14,503,603
<500 employees
1,049
1,100
44,594
20,970,635
500 + employees
130
344
120,494
128,313,895
Total
1,179
1,444
165,088
149,284,530
As defined in section 1192(e)(3)(B)(i) of the Act, a selected drug must have total expenditures of $200 million or more during the defined measurement period, between June 1, 2022 and May 31, 2023, for initial price applicability year 2026, adjusted by inflation in subsequent years (hereinafter the “low-spend Medicare drug exclusion”).
For purposes of this proposed analysis, we are unable to determine the actual employee size or revenue of all manufacturers that may ever have a selected drug in the Negotiation Program. Assuming the low-spend Medicare drug exclusion described previously, we estimate that no Primary Manufacturer would be considered to be a small entity because we expect that the resources needed to produce and distribute a drug with Medicare expenditures that meet this threshold would require a workforce larger than the small entity threshold of 1,300 employees. As Table 22 demonstrates, none of the drugs that were selected for negotiation, as set forth in § 429.105 of this proposed rule, were manufactured by small entities during the first 3 years of the Negotiation Program.
Table 22—Percent of Small Entity Primary Manufacturers With Selected Drugs During Initial Price Applicability Years 2026-2028 85
Initial price applicability year
Company size
Total number of employees in the U.S.
Percent of
unique
companies (%)
2026
Small
1,300 or fewer
0
Large
1,300+
100
2027
Small
1,300 or fewer
0
Large
1,300+
100
2028
Small
1,300 or fewer
0
Large
1,300+
100
2. Description of the Potential Impacts on Small Entities
With the expiration of the Small Biotech Exception, which applies to initial price applicability years 2026, 2027, and 2028, and the implementation of the Temporary Floor for Small Biotech Drugs, which becomes effective for initial price applicability years 2029 and 2030, manufacturers of small biotech drugs could have a selected drug for initial price applicability year 2029 or a future year. It is possible that a manufacturer of a small biotech drug could have 1,300 or fewer employees, but CMS does not have credible information to forecast if or when such manufacturers would have a selected drug.
CMS notes that the estimates of manufacturer size and impact of this rule are based on available data which could change in the future and as such, the estimated impacts could vary. Specifically, the estimates are based on the current status of drug negotiations, employment and revenue information using 2024 or other available data as of the publication of this proposed rule. Despite this uncertainty, CMS certifies that the proposed rule, if finalized as proposed, would not have a significant impact on a substantial number of small entity Primary Manufacturers because the resources needed to produce and distribute a drug with expenditures that meet the low-spend Medicare drug exclusion is expected to require a workforce larger than the small entity threshold, therefore, we expect that no Primary Manufacturer would qualify as a small entity. A timeline of potential impacts to small entities in the Negotiation Program are showed in Table 23. CMS welcomes comments on
( printed page 36336)
our conclusion, approach, assumptions, and data used to estimate these impacts.
Table 23—Potential Impacts to Small Entities in the Negotiation Program
2026
2027
2028
2029
2030
Primary Manufacturers
No changes
No changes
No changes
Small Biotech Exception no longer applies; previously excepted small biotech drugs now eligible for Negotiation Program
Small Biotech Exception no longer applies; previously excepted small biotech drugs now eligible for Negotiation Program.
3. Alternatives To Minimize the Impact on Small Entities
The vast majority of the policies discussed in this proposed rule, including timing for reporting and publications and effective dates of rates, are required by sections 1191 through 1198 of the Act. Due to the IRA establishing the requirements for the selection and identification of negotiation-eligible drugs and the requirements for the negotiation or renegotiation process rather than this proposed rule, CMS is unable to propose alternatives that will accomplish the purpose of the Negotiation Program in a manner consistent with law. In addition, to the extent practicable, processes were standardized and clarified to improve efficiency for Primary Manufacturers to comply with program requirements.
G. Unfunded Mandates Reform Act (UMRA)
Section 202 of UMRA also requires that agencies assess anticipated costs and benefits before issuing any rule whose mandates require spending in any 1 year of $100 million in 1995 dollars, updated annually for inflation. In 2026, that threshold is approximately $193 million. This proposed rule is not anticipated to have an unfunded effect on State, local, or Tribal governments, in the aggregate, or on the private sector of $193 million or more. The analysis of impacts on this proposed rule does not report an estimate of any unfunded effect on State, local, or Tribal governments, in the aggregate, or on the private sector that exceeds the $193 million threshold. However, this proposed rule, if finalized as proposed, would result in additional impacts associated with changes in behavior; these are not quantified. Therefore, the Secretary has concluded that the requirements of section 202 of the UMRA have been met for this proposed rule. We request comments, including on the potential magnitude of this impact.
H. Federalism
Executive Order 13132
establishes certain requirements that an agency must meet when it promulgates a rule that imposes substantial direct requirement costs on State and local governments, preempts State law, or otherwise has federalism implications. Since this proposed rule does not impose any substantial costs on State or local governments, preempt State law or have federalism implications, the requirements of
Executive Order 13132
are not applicable.
Executive Order
14192, titled “Unleashing Prosperity Through Deregulation” was issued on January 31, 2025, and requires that “any new incremental costs associated with new regulations shall, to the extent permitted by law, be offset by the elimination of existing costs associated with at least 10 prior regulations.”
J. Conclusion
The analysis in the previous sections, together with the remainder of this preamble, provided an initial Regulatory Flexibility Analysis. The previous analysis, together with the preceding portion of this preamble, provides an RIA. In accordance with the provisions of Executive Order 12866, this regulation was reviewed by the Office of Management and Budget.
VI. Response to Comments
Because of the large number of public comments we normally receive on
Federal Register
documents, we are not able to acknowledge or respond to them individually. We will consider all comments we receive by the date and time specified in the
DATES
section of this preamble, and, when we proceed with a subsequent document, we will respond to the comments in the preamble to that document.
Mehmet Oz, Administrator of the Centers for Medicare & Medicaid Services, approved this document on May 26, 2026.
2. Section 423.100 is amended by revising the definition of “Corresponding drug” and revising and republishing the definition of “Negotiated price” to read as follows:
Corresponding drug
means, respectively, a generic or authorized generic of a brand name drug, an interchangeable biological product of a reference product, or an unbranded biological product marketed under the same biologics license application (BLA) as a brand name biological product. A corresponding drug does not include a selected drug.
* * * * *
Negotiated price
means the price for a covered Part D drug that meets all of the following:
(1) The Part D sponsor (or other intermediary contracting organization) and the network dispensing pharmacy or other network dispensing provider have negotiated as the lowest possible reimbursement such network entity will receive, in total, for a particular drug;
( printed page 36337)
(2) Subject to paragraph (4) of this definition, meets all of the following:
(i) Includes all price concessions (as defined in this section) from network pharmacies or other network providers.
(ii) Includes any dispensing fees.
(iii) Includes any applicable sales tax.
(iv) Includes any applicable vaccine administration fee.
(v) Excludes additional contingent amounts, such as incentive fees, if these amounts increase prices.
(3) Is reduced by non-pharmacy price concessions and other direct or indirect remuneration that the Part D sponsor passes through to Part D enrollees at the point of sale.
(4) For a covered Part D drug that is a selected drug and for each year of a price applicability period with respect to such selected drug, the negotiated price used for payment, as defined in paragraphs (1) through (3) of this definition, must not exceed the maximum fair price, as defined at section 1191(c)(3) of the Act, for such drug, plus any applicable dispensing fees.
Requirements related to qualified prescription drug coverage.
* * * * *
(b) * * *
(2) * * *
(vii) Except as specified in paragraph (viii) of this section, for 2026 and each subsequent year, include each Part D drug that is a selected drug under section 1192 of the Act for which a maximum fair price (as defined in section 1191(c)(3) of the Act) is in effect with respect to the year.
(viii) Nothing in paragraph (b)(2)(vii) of this section may be construed as prohibiting a Part D plan sponsor from removing such a selected drug from its formulary if such removal meets the requirements specified in paragraph (e)(2)(i) of this section and the notice requirements specified in paragraphs (f)(2), (3), and (4) of this section.
(1) This part is based on the indicated provision of the following sections of the Act:
1191. Establishment of the Program.
1192. Selection of Negotiation-Eligible Drugs as Selected Drugs.
1193. Manufacturer Agreements.
1194. Negotiation and Renegotiation Process.
1195. Publication of Maximum Fair Prices.
1196. Administrative Duties and Compliance Monitoring.
1197. Civil Monetary Penalties.
1198. Limitation on Administrative and Judicial Review.
(2) The following specific sections of the Inflation Reduction Act also address the Medicare Drug Price Negotiation Program:
11001. Providing for Lower Prices for Certain High-Priced Single Source Drugs.
11002. Special Rule to Delay Selection and Negotiation of Biologics for Biosimilar Market Entry.
(b)
Scope.
This part sets forth the requirements of the Medicare Drug Price Negotiation Program, which requires the Secretary to negotiate and renegotiate, for applicable periods, Medicare prices for certain high expenditure, single source drugs and biological products.
(c)
Severability.
Were any provision of this part to be held invalid or unenforceable by its terms, or as applied to any person or circumstance, such provision would be severable from this part and the invalidity or unenforceability would not affect the remainder thereof or any other part of
( printed page 36338)
this subchapter or the application of such provision to other persons not similarly situated or to other, dissimilar circumstances.
As used in this part, unless otherwise specified, the following definitions apply:
Additional delay request
means a request to delay the inclusion on the selected drug list of a reference drug for which an initial delay request has been granted for a second initial price applicability year consistent with § 429.110 and section 1192(f)(1)(B)(i)(II) of the Act.
Applicable program agreement
means an agreement under the Manufacturer Discount Program as specified in section 1860D-14C of the Act or a rebate agreement described in section 1927(b) of the Act.
Authorized generic drug
means a drug as defined in section 505(t)(3) of the Federal Food, Drug, and Cosmetic (FD&C) Act and a biological product that has been licensed under section 351(a) of the Public Health Service (PHS) Act and is marketed, sold, or distributed directly or indirectly to the retail class of trade under a different labeling, packaging (other than repackaging as the reference product in blister packs, unit doses, or similar packaging for institutions), product code, labeler code, trade name, or trademark than the reference product.
Authorized representative
means an individual that has the authority or capacity to legally bind the Primary Manufacturer to the terms and conditions of the Negotiation Program Agreement and meets one of the following criteria:
(1) Chief Executive Officer of the Primary Manufacturer.
(2) Chief Financial Officer of the Primary Manufacturer.
(3) An individual with equivalent authority to a Chief Executive Officer or Chief Financial Officer of the Primary Manufacturer.
(4) An individual that has been granted delegation of signature authority on behalf of one of the individuals specified in paragraphs (1) through (3) of this definition.
Average manufacturer price (AMP)
has the meaning set forth in section 1927(k)(1) of the Act.
Average non-Federal average manufacturer price (non-FAMP)
has the meaning set forth in section 1194(c)(6) of the Act.
Average sales price (ASP)
means the manufacturer's price for a quarter for a drug represented by a particular 11-digit National Drug Code (NDC-11) determined under 42 CFR 414.804 of this chapter and as reported in section 1927(b)(3) of the Act.
Billing unit
means the identifiable quantity of a drug or biological product associated with a billing and payment code (for example, a Healthcare Common Procedure Coding System code), as established by CMS.
Biologics License Application (BLA)
means an application submitted under section 351 of the PHS Act.
Biosimilar biological product or biosimilar
has the meaning set forth in section 1847A(c)(6) of the Act.
Biosimilar Delay Request
means an Initial Delay Request or an Additional Delay Request.
Biosimilar Manufacturer
means one of the following:
(1) The BLA holder for the Biosimilar;
(2) If a BLA has been submitted to the FDA for review but the Biosimilar has not been licensed, the sponsor of the BLA submitted for review by the FDA; or
(3) If the Biosimilar has not been licensed and the BLA has not been submitted to the FDA, the organization planning to be the sponsor when the BLA is submitted for review by the FDA.
BLA holder
means the entity that is the holder of the license(s) permitting marketing of a biological product in accordance with section 351 of the PHS Act.
Bona Fide Marketing
means that an approved generic drug or licensed biosimilar is marketed as determined by CMS based on the information considered in § 429.130(a).
Combined Part B and Part D amount
means an amount equal to the weighted average of the payment amount under section 1847A(b)(4) of the Act and the sum of the plan-specific enrollment weighted amount as determined by CMS under § 429.410(c).
CPI-U
means the monthly Consumer Price Index for All Urban Consumers (United States city average) index level for all items from the Bureau of Labor Statistics.
Direct and Indirect Remuneration (DIR)
has the meaning set forth in § 423.308 of this chapter.
Drug covered under Part D
means a covered Part D drug as defined in section 1860D-2(e) of the Act.
Drug payable under Part B
means a drug or biological product for which payment may be made under part B of Title XVIII of the Act.
Estimated remuneration at point-of-sale amounts (ERPOSA)
means the estimated amount of rebates or other price concessions that the Part D plan sponsor is required to apply, or has elected to apply, to the negotiated price as a reduction in the drug price made available to the beneficiary at the point of sale.
Extended-monopoly drug
has the meaning set forth in section 1194(c)(4) of the Act.
FDA-approved indication
means the information included in drug labeling per 21 CFR 201.57(c)(2) or FDA regulations as applicable.
Fixed combination drug
has the meaning set forth in 21 CFR 300.50.
Generic drug
means a drug approved in an Abbreviated New Drug Application (ANDA) under section 505(j) of the FD&C Act.
Healthcare Common Procedure Coding System (HCPCS) code
means a billing and payment code, as established by CMS for payment under Part B, used to describe a drug or biological and for which CMS may publish a payment amount.
High Likelihood Deadline
means the date that is 2 years after the statutorily defined selected drug publication date for the initial price applicability year for which a reference drug would be included on the selected drug list absent a successful Initial Delay Request.
Initial Delay Period
means the time period between the selected drug publication date—
(1) For the initial price applicability year for which the Reference Drug otherwise would have been included on the selected drug list but for the successful Initial Delay Request, as proposed in § 429.110(g); and
(2) With respect to the initial price applicability year that is 1 year after the initial price applicability year for which the Reference Drug otherwise would have been included on the selected drug list but for the successful Initial Delay Request as set forth in section 1192(f)(2) of the Act.
Initial Delay Request
means a request to delay the inclusion of a reference drug on the selected drug list by one initial price applicability year consistent with § 429.110(c) of this part and section 1192(f)(1)(B)(i)(I) of the Act.
Initial price applicability year
has the meaning set forth in section 1191(b)(1) of the Act.
Long-monopoly drug
has the meaning set forth in section 1194(c)(5) of the Act.
Manufacturer
has the meaning set forth in section 1191(c)(1) of the Act.
Manufacturer Discount Program
means the Medicare Part D Manufacturer Discount Program established under section 1860D-14C of the Act.
( printed page 36339)
Maximum fair price (MFP)
has the meaning set forth in section 1191(c)(3) of the Act.
Medicare Drug Price Negotiation Program (or Negotiation Program)
means the program created by sections 11001 and 11002 of the Inflation Reduction Act and codified in sections 1191 through 1198 of the Act and as amended.
Medicare Drug Price Negotiation Program Agreement (or Negotiation Program Agreement)
means the agreement between a Primary Manufacturer and CMS as set forth in § 429.200 and section 1193(a) of the Act.
NDA holder
means the entity that is the holder of the approval(s) to market a drug product in accordance with section 505(c) of the FD&C Act.
Negotiation-eligible drug
has the meaning set forth in section 1192(d) of the Act and is identified in accordance with § 429.115.
Negotiation period
has the meaning set forth in section 1191(b)(4) of the Act.
Net Part D Plan Payment and Beneficiary Liability
means, for purposes of the Medicare Drug Price Negotiation Program, the total gross covered prescription drug cost for a selected drug covered under Part D net of direct and indirect remuneration (DIR) and Manufacturer Discount Program payments and excluding prescription drug event (PDE) records for which a compound code indicates the PDE record is for a compounded drug.
New Drug Application (NDA)
means an application submitted under section 505(b) of the FD&C Act.
Off-label use
means use for a condition for a selected drug or a therapeutic alternative that is not an FDA-approved indication but is included in evidence-based clinical practice guidelines and is a medically accepted indication payable under Part B or covered under Part D or both, taking into consideration major drug compendia, authoritative medical literature, accepted standards of medical practice, or some combination thereof.
Outcomes
means the impact of an intervention, which may be clinical or related to the functioning, symptoms, quality of life, or other aspects of a patient's life.
Part B data
means Original Medicare (OM) Part B claims data and Medicare Advantage (MA) encounter data for Part B items or services.
Partnership
has the meaning set forth in section 1192(f)(1)(C)(ii) of the Act.
Personally identifiable information (PII)
has the meaning set forth at 2 CFR 200.1.
Plasma-derived product
has the meaning set forth in section 1192(e)(3)(C) of the Act.
Preliminary price
means the numerical dollar amount used by CMS in developing an initial offer in accordance with § 429.510(e) by adjusting the starting point of a selected drug based on section 1194(e)(2) factors.
Price applicability period
has the meaning set forth in section 1191(b)(2) of the Act.
Primary Manufacturer
means the manufacturer identified by CMS as the NDA holder or the BLA holder for the selected drug.
Private label distributor
has the meaning set forth in 21 CFR 207.1.
Protected health information (PHI)
has the meaning set forth at 45 CFR 160.103.
Qualifying single source drug
has the meaning set forth in section 1192(e) of the Act and is identified in accordance with § 429.125.
Rare disease or condition
has the meaning set forth in section 526(a)(2) of the FD&C Act.
Reference Drug
means a negotiation-eligible drug that includes the reference product for a Biosimilar that is named in a Biosimilar Delay Request.
Reference Manufacturer
means the Primary Manufacturer of the Reference Drug that is named in a Biosimilar Delay Request.
Reference Product
has the meaning set forth in section 1191(c)(4) of the Act.
Relabeler
has the meaning set forth in 21 CFR 207.1.
Renegotiation-eligible drug
has the meaning set forth in section 1194(f)(2) of the Act.
Repackager
has the meaning given the term “repacker” set forth in 21 CFR 207.1.
Request to Terminate
means a written request submitted by a Primary Manufacturer to CMS, that CMS determines meets the conditions described in § 429.205(b)(1), to request termination of its applicable program agreements in the context of a Primary Manufacturer's decision not to enter into or to terminate a Negotiation Program Agreement.
Secondary Manufacturer
means a manufacturer of a drug product included in the selected drug, that is not the Primary Manufacturer for the selected drug, and that either—
(1) Is listed as a manufacturer in an NDA or BLA for the selected drug; or
(2) Markets the selected drug in accordance with an agreement with the Primary Manufacturer but is not listed on an NDA or BLA of the selected drug.
A Secondary Manufacturer includes any manufacturer of any authorized generic drug(s) and any repackager or relabeler of the selected drug that meets either of these criteria.
Second Delay Period
means the time period between the publication date of the selected drug list for initial price applicability year that is—
(1) One year after the initial price applicability year for which the Reference Drug would have been included on the selected drug list but for the successful Initial Delay Request; and
(2) Two years after the initial price applicability year for which the Reference Drug would have been included on the selected drug list but for the successful Initial Delay Request.
Section 1194(e)(1) factors
mean the factors described in section 1194(e)(1) of the Act.
Section 1194(e)(2) factors
mean the factors described in section 1194(e)(2) of the Act.
Selected drug
has the meaning set forth in section 1192(c) of the Act and is identified in accordance with § 429.105.
Selected drug publication date
has the meaning set forth in section 1191(b)(3) of the Act.
Self-administered drug
means a drug or biological identified as self-administered by the U.S. Department of Health and Human Services Office of Inspector General (OIG) in accordance with section 1847A(g)(1) of the Act.
Sequestration payment adjustment
means, when applicable, the amount that is applied to a Part B claim to determine the Medicare payment amount—after determining coinsurance, deductible, merit-based incentive payment adjustments, and any applicable Medicare Secondary Payment adjustments.
Small Biotech Drug
means a drug that is determined by CMS under § 429.440(b)(2), in accordance with section 1192(d)(2) of the Act, as eligible for the Temporary Floor for Small Biotech Drugs.
Specified Manufacturer
has the meaning set forth in section 1860D-14C(g)(4)(B)(ii) of the Act, as determined by CMS for the purposes of the Manufacturer Discount Program in accordance with 42 CFR 423.2716, 423.2720, and 423.2724.
Starting point
means the numerical dollar amount used by CMS in developing an initial offer in accordance with § 429.510(d) that is then adjusted by CMS based on section 1194(e)(2) factors to determine the preliminary price, per the process described in § 429.510(e).
( printed page 36340)
Temporary Floor for Small Biotech Drugs
has the meaning set forth in § 429.440(b)(3).
Therapeutic advance
means a demonstrated improvement in one or more outcomes or other clinical considerations for an identified condition of a selected drug as compared to its therapeutic alternative(s), if any. For the purpose of developing the initial offer CMS considers therapeutic advance as of the date specified in § 429.505(d)(2). For purposes of the Negotiation Program, anytime CMS considers an unmet medical need, CMS will consider the extent to which the drug addresses an unmet medical need at the time of consideration based on all available information at such time of consideration.
Therapeutic alternative
means a pharmaceutical product or group of pharmaceutical products other than the selected drug that may be used to treat the same condition or disease state as the selected drug.
Total allowed charges
means the amount that is inclusive of the beneficiary coinsurance and Medicare payment for the covered Part B item or service paid for under part B of Title XVIII of the Act, without a sequestration payment adjustment applied.
Total expenditures
has the meaning set forth in section 1191(c)(5) of the Act, as determined under § 429.120.
Total expenditures measurement period
means the 12-month period ending on October 31 of the year prior to the year of the selected drug publication date with respect to an initial price applicability year.
Total gross covered prescription drug costs
has the meaning given the term “gross covered prescription drug costs” set forth in 42 CFR 423.308 of this chapter.
Unit
has the meaning set forth in section 1191(c)(6) of the Act.
Unmet medical need
means a circumstance in which the relevant disease or condition is one for which no other treatment options exist, or existing treatments do not adequately address the disease or condition. For purposes of the Negotiation Program, anytime CMS considers an unmet medical need, CMS would consider the extent to which the drug addresses an unmet medical need at the time of consideration based on all available information at such time of consideration.
Wholesale Acquisition Cost (WAC) unit price
means the manufacturer's list price for the drug or biological product to wholesalers or direct purchasers in the United States, not including prompt pay or other discounts, rebates or reductions in price, for the most recent month for which the information is available, as reported in wholesale price guides or other publications of drug or biological product pricing data (as defined in section 1847A(c)(6)(B) of the Act). The WAC unit price is reported at the NDC-11 level.
(a) There is no administrative or judicial review of any of the following:
(1) The determination of a unit, with respect to a drug or biological product, under section 1191(c)(6) of the Act.
(2) The selection of drugs under section 1192(b) of the Act, the determination of negotiation-eligible drugs under section 1192(d) of the Act, the determination of qualifying single source drugs under section 1192(e) of the Act, and the application of section 1192(f) of the Act.
(3) The determination of a maximum fair price under subsection (b) or (f) of section 1194 of the Act.
(4) The determination of renegotiation-eligible drugs under section 1194(f)(2) of the Act and the selection of renegotiation-eligible drugs under section 1194(f)(3) of the Act.
(a)
General.
Each drug included on the selected drug list for an initial price applicability year is a selected drug with respect to such initial price applicability year and each subsequent year unless and until CMS makes a determination in accordance with § 429.135(a).
(b) With respect to each initial price applicability year beginning with initial price applicability year 2029, CMS publishes a list with the following information no later than the selected drug publication date as defined in § 429.20 with respect to such initial price applicability year:
(1) The selected drug list, which includes the 20 (or all, if such number is less than 20) selected drugs with respect to such initial price applicability year, determined in accordance with § 429.105(c).
(2) The drugs selected for renegotiation, if any, determined in accordance with § 429.610.
(3) For each selected drug included on the selected drug list published in accordance with paragraph (b)(1) of this section, CMS adds the following information to the MFP file:
(i) The active moiety/active ingredient/antigen component (or the distinct combination thereof), identified in accordance with § 429.125(b).
(ii) The NDC-11s identified in accordance with paragraph (c)(1) of this section, and the corresponding NDC-9s and HCPCS codes (as applicable).
(4) For each drug selected for renegotiation published in accordance with paragraph (b)(2) of this section, if any, CMS adds the following information to the MFP file:
(i) The active moiety/active ingredient/antigen component (or the distinct combination thereof) previously identified for the initial price applicability year for which the drug was originally selected for negotiation.
(ii) The NDC-11s identified in accordance with paragraph (c)(1) of this section as updated to reflect information previously submitted by the Primary Manufacturer, including submissions in accordance with this section, and the corresponding NDC-9s and HCPCS codes (as applicable).
(c)
Identification of the list of NDC-11s of the selected drug.
CMS identifies a list of the NDCs of each selected drug, including for a drug selected for renegotiation, that is developed and maintained using the following process:
(1) CMS identifies a baseline list of NDC-11s associated with the NDA(s)/BLA(s) of the selected drug.
(2) CMS transmits the NDC-11s identified in paragraph (c)(1) of this section to the Primary Manufacturer.
(3) CMS revises its list of NDC-11s of each selected drug, as appropriate, including without limitation using information submitted by the Primary Manufacturer in accordance with this section.
(d)
Primary Manufacturer submission of information for CMS' list of NDC-11s.
In accordance with the requirements of the Negotiation Program Agreement as set forth in § 429.200, including to ensure CMS' list of NDC-11s of the selected drug developed in accordance with § 429.100(c) is complete and accurate, the Primary Manufacturer must submit all of the following information to CMS in a form and manner specified by CMS by 11:59 p.m. PST on March 1 of the calendar year of the selected drug publication date for the selected drug, inclusive of NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer:
(1) Any NDC-11s associated with the NDA(s)/BLA(s) of the selected drug that do not appear on CMS' list of NDC-11s of the selected drug, including any missing NDC-11s of a Secondary Manufacturer.
(2) Any NDC-11s on CMS' list of NDC-11s of the selected drug that are
( printed page 36341)
distributed by or under the name of a private label distributor.
(3) Any NDC-11s on CMS' list of NDC-11s of the selected drug that are not manufactured, marketed, controlled or sold by the Primary Manufacturer or a Secondary Manufacturer.
(4) Any NDC-11s on CMS' list of NDC-11s of the selected drug that represent a sample package.
(5) Any NDC-11s on CMS' list of NDC-11s of the selected drug that represent an inner package or an outer package.
(6) Any NDC-11s on CMS' list of NDC-11s of the selected drug that have been discontinued and the discontinuation date for each.
(e)
Primary Manufacturer requirement to timely report changes.
In accordance with the requirements of the Negotiation Program Agreement as set forth in § 429.200, including to ensure CMS' list of NDC-11s of the selected drug developed in accordance with § 429.100(c) remains complete and accurate, the Primary Manufacturer must update the information provided to CMS under paragraph (d) of this section, in a form and manner specified by CMS, at least 30 calendar days prior to the change taking effect.
(f) CMS' list of NDC-11s of the selected drug identified in accordance with paragraph (c) of this section is used in the administration of the Negotiation Program, including all of the following:
(1) To identify the NDC-11s of the selected drug that are subject to the negotiation process set forth in subpart F of this part and the renegotiation process set forth in subpart G of this part, as applicable.
(2) To identify the NDC-11s of the selected drug to which the MFP applies for the price applicability period.
(3) To calculate the ceiling set forth in § 429.410(b) for drugs selected for negotiation.
(4) To calculate the ceiling set forth in § 429.620(b) for drugs selected for renegotiation.
(5) To calculate how to apply the MFP across dosage forms and strengths as set forth in § 429.700 for selected drugs and in § 429.600(b)(2) for drugs selected for renegotiation.
(a) With respect to each initial price applicability year beginning with initial price applicability year 2029, CMS—
(1) Ranks the list of negotiation-eligible drugs identified in § 429.115 by combined total expenditures under both Part B and Part D, calculated using the methodology set forth at § 429.120; and
(2) Lists the ranked drugs in descending order, with the negotiation-eligible drug with the highest combined total expenditures under Part B and Part D listed first and the negotiation-eligible drug with the lowest combined total expenditures under Part B and Part D listed last.
(b) CMS removes from the ranked list of negotiation-eligible drugs determined in accordance with paragraph (a) of this section any biological products that qualify for delayed selection as determined in accordance with the biosimilar delay process set forth in § 429.110.
(c) With respect to each initial price applicability year beginning with initial price applicability year 2029, CMS selects for negotiation the 20 highest ranked (or all, if the number is less than 20) negotiation-eligible drugs remaining on the ranked list of negotiation-eligible drugs determined in accordance with paragraph (a) of this section and subject to paragraph (b) of this section. If two or more negotiation-eligible drugs have the same combined total expenditures under Parts B and D to the cent, and such combined total expenditures are the 20th highest (or the highest, if the number is less than 20) among negotiation-eligible drugs, CMS ranks those negotiation-eligible drugs based on which drug has the earlier approval or licensure date (as applicable) identified at § 429.125(c), and selects from among the drugs with the same combined total expenditures based on earliest approval or licensure date until there are 20 selected drugs (or until all drugs are selected, if the number of negotiation-eligible drugs is less than 20).
(a) For purposes of this section, all references to “marketed” or “marketing” mean Bona Fide Marketing as defined in § 429.20 and set forth at § 429.130(a).
(b) General. A Biosimilar Manufacturer may submit a request, prior to the selected drug publication date, to delay the inclusion of a Reference Drug from the selected drug list for either of the following:
(1) One initial price applicability year based on the criteria specified in paragraph (c) of this section.
(2) A second initial price applicability year after such first initial price applicability year based on the criteria specified in paragraph (e) of this section.
(c)
First 1-year delay (Initial Delay Request).
To delay the inclusion of a Reference Drug for one initial price applicability year, CMS must determine the following:
(1) The Initial Delay Request meets all of the following criteria:
(i) The Reference Drug would be an extended-monopoly drug included on the selected drug list for the initial price applicability year, absent the Biosimilar Delay.
(ii) The Reference Drug includes the reference product identified in the Biosimilar's application for licensure under section 351(k) of the PHS Act that has either been—
(A) Approved by the FDA; or
(B) Accepted for review by the FDA.
(iii) If the Biosimilar has been licensed by FDA, no more than 1 year has elapsed since the licensure of the Biosimilar under section 351(k) of the PHS Act and marketing for the Biosimilar has not commenced.
(iv) The Biosimilar Manufacturer and the Reference Manufacturer—
(A) Are not the same manufacturer and must not be treated as the same manufacturer, where all persons treated as a single employer under subsection (a) or (b) of section 52 of the IRC, or in a Partnership, must be treated as one manufacturer; and
(B) Have not entered into any agreement that—
(1)
Requires or incentivizes the Biosimilar Manufacturer to submit a Biosimilar Delay Request; or
(2)
Directly or indirectly restricts the quantity of the Biosimilar that may be sold in the United States on or after the selected drug publication date of the applicable initial price applicability year.
(2) If the requirements in paragraph (c)(1) of this section are met, that there is a high likelihood, as specified in paragraph (d) of this section, that the Biosimilar will be licensed and marketed before the High Likelihood Deadline.
(d)
High likelihood.
There is a high likelihood the Biosimilar will be licensed and marketed before the High Likelihood Deadline if each of the following criteria are met:
(1) By no later than a date to be specified by CMS, which will be in January in the calendar year of the selected drug publication date for the initial price applicability year for which the Biosimilar Manufacturer requests the delay, an application for licensure under section 351(k) of the PHS Act for the Biosimilar has been either—
(i) Approved by the FDA; or
(ii) Accepted for review by FDA.
(2) CMS has identified clear and convincing evidence that the Biosimilar will be marketed before the High Likelihood Deadline through the following circumstances:
(i) Patents related to the Reference Drug are unlikely to prevent the
( printed page 36342)
Biosimilar from being marketed before the High Likelihood Deadline, as CMS identifies within the documentation specified in paragraph (f)(1) of this section, meaning one or more of the following:
(A) There will be no unexpired patents relating to the reference product included in the Reference Drug that are applicable to the Biosimilar.
(B) One or more court decisions or decisions by the United States Patent and Trademark Office (USPTO)'s Patent Trial and Appeal Board (PTAB) establish the invalidity, unenforceability, or non-infringement of any potentially applicable unexpired patents relating to the reference product included in the Reference Drug that the patent holder asserted was applicable to the Biosimilar.
(C) Neither a court nor PTAB has adversely ruled against the Biosimilar Manufacturer's patent assertion(s) pertaining to an unexpired patent or patent(s) relating to the reference product included in the Reference Drug applicable to the Biosimilar and the Biosimilar Manufacturer has publicly announced a precise launch date for the Biosimilar that is a calendar date before the High Likelihood Deadline.
(D) The Biosimilar Manufacturer has a signed agreement with the Reference Manufacturer that permits the Biosimilar Manufacturer to market the Biosimilar before the High Likelihood Deadline, without imposing improper constraints, that may include, but are not limited to, the circumstances listed in paragraphs (c)(1)(iv)(A), (B)(
1), and (
2) of this section (which prohibits the Biosimilar Manufacturer and the Reference Manufacturer being the same or being treated as the same, requiring or incentivizing the submission of a Biosimilar Delay Request, or directly or indirectly restricting the quantity of the Biosimilar sold in the United States on or after the selected drug publication date of the applicable initial price applicability year), on the Biosimilar Manufacturer.
(ii) The Biosimilar Manufacturer will be operationally ready to market the Biosimilar before the High Likelihood Deadline, as CMS identifies within the documentation specified in paragraph (f)(1) of this section, meaning the Biosimilar Manufacturer is taking the actions, activities, and milestones that are typical of the normal course of business leading up to the marketing of the Biosimilar.
(e)
Second 1-year delay (Additional Delay Request).
To delay the inclusion of a Reference Drug for a second initial price applicability year, CMS must determine the following:
(1) The Additional Delay Request meets all of the following criteria:
(i) The Biosimilar listed in the Additional Delay Request was identified in a successfully granted Initial Delay Request (consistent with paragraph (c) of this section).
(ii) During the time between the publication date of the selected drug list for the initial price applicability year for which the Initial Delay Request was granted and the date that is 1 year following that publication date, under section 351(k) of the PHS Act, the Biosimilar has not been both—
(A) Licensed; and
(B) Marketed.
(iii) The Additional Delay Request meets the requirements set forth in paragraphs (c)(1)(ii) through (iv) of this section.
(iv) The Reference Drug would not be a long-monopoly drug (as set forth in § 429.20) for the initial price applicability year for which the Biosimilar Manufacturer is submitting the Additional Delay Request.
(2) If the requirements in paragraph (e)(1) of this section are met, CMS must reevaluate and determine that there is a high likelihood, as specified in paragraph (d) of this section, that the Biosimilar will be licensed and marketed before the High Likelihood Deadline.
(3) If the requirements in paragraph (e)(2) of this section are met, CMS must determine based on a holistic review of the Additional Delay Request, including within the documentation specified in paragraph (f) of this section, there is clear and convincing evidence that the Biosimilar Manufacturer has made a significant amount of progress towards licensure and marketing of the Biosimilar since the Biosimilar Manufacturer's submission of the successful Initial Delay Request.
(f)
Biosimilar delay request submission.
A Biosimilar Manufacturer may submit a Biosimilar Delay Request to CMS in a form and manner specified by CMS, by no later than a date to be specified by CMS, which will be no later than the end of December of the calendar year prior to the selected drug publication date for such initial price applicability year for which the Biosimilar Manufacturer may submit a Biosimilar Delay Request, and must provide the information specified in paragraphs (1) through (3) of this paragraph (f), as applicable:
(1) With the Biosimilar Manufacturer's Initial Delay Request or Additional Delay Request to support the determinations specified in paragraphs (c) and (e) of this section, as applicable:
(i) All agreements related to the Biosimilar filed with the Federal Trade Commission or the Assistant Attorney General under section 1112(a) and (c) of the Medicare Prescription Drug, Improvement, and Modernization Act of 2003;
(ii) To the extent available, the manufacturing schedule for the Biosimilar submitted to FDA during the review of the licensure application under section 351(k) of the PHS Act.
(iii) To the extent available, disclosures that pertain to the marketing of the Biosimilar (for example, in filings by the Biosimilar Manufacturer of the Biosimilar with the Securities and Exchange Commission required under section 12(b), 12(g), 13(a), or 15(d) of the Securities Exchange Act of 1934 or comparable documentation that is distributed to shareholders of privately held companies) about capital investment, revenue expectations, and actions taken by the manufacturer that are typical of the normal course of business in the year (or the 2 years, as applicable) before the marketing of a Biosimilar;
(2) Upon request from CMS after review of a Biosimilar Delay Request, including the information provided consistent with paragraph (f)(1) of this section, additional information and documents from the Biosimilar Manufacturer to be submitted in a form and manner specified by CMS as necessary for CMS to make a determination for an Initial Delay Request or an Additional Delay Request, as applicable.
(3) At the Biosimilar Manufacturer's option, after the deadline for submitting a Biosimilar Delay Request specified in paragraph (f) of this section but prior to the selected drug publication date for such initial price applicability year for which the Biosimilar Manufacturer may submit a Biosimilar Delay Request, updates on the status of the Biosimilar's application for licensure in a form and manner specified by CMS by no later than a date to be specified by CMS.
(g)
Notice of determinations.
(1) After review of a Biosimilar Delay Request, CMS provides in writing:
(i) A notice of determination, on or after the selected drug publication date for the applicable initial price applicability year, at a time specified by CMS, to:
(A) The Biosimilar Manufacturer that submitted a request, which includes:
(1)
Whether the Biosimilar Delay Request was successful or unsuccessful; and
(2)
If unsuccessful, the reason CMS determined the request was unsuccessful.
( printed page 36343)
(B) If a Biosimilar Delay Request is successful, the Reference Manufacturer of the reference product named in a successful Biosimilar Delay Request.
(2) CMS publishes the number of Reference Drugs that would have been selected drugs for the applicable initial price applicability year absent the successful Biosimilar Delay Requests as part of publishing the selected drug list (as set forth in § 429.100).
(3) After review for licensure and marketing consistent with paragraph (h) of this section, CMS provides in writing by a date to be specified by CMS, which will be no later than the end of October in the calendar year of the selected drug publication date for the initial price applicability year for which CMS determined the Biosimilar Manufacturer's Biosimilar Delay Request successful, a notice of determination to the Biosimilar Manufacturer of CMS' determination as to whether the Biosimilar is or is not licensed and marketed during the Initial Delay Period or the Second Delay Period, as applicable.
(h)
Review for failure of the biosimilar to be licensed and marketed
—
(1) CMS determines whether a Biosimilar named in a successful Initial Delay Request has been licensed and marketed during the Initial Delay Period.
(i) If CMS determines that the Biosimilar has not been licensed and marketed during the Initial Delay Period, the Biosimilar Manufacturer may submit an Additional Delay Request consistent with paragraph (e) of this section.
(ii) Subject to the circumstance in paragraph (h)(3) of this section, CMS includes the Reference Drug on the selected drug list for the initial price applicability year that is 1 year after the initial price applicability year for which the Reference Drug would have been included on the selected drug list if not for the successful Initial Delay Request if either of the following occurs:
(A) The Biosimilar Manufacturer does not submit an Additional Delay Request for the Biosimilar included in the successful Initial Delay Request.
(B) CMS determines that the Additional Delay Request submitted by the Biosimilar Manufacturer for the Biosimilar named in the successful Initial Delay Request does not meet the requirements in paragraph (e) of this section.
(2) CMS determines whether a Biosimilar named in a successful Additional Delay Request has been licensed and marketed during the Second Delay Period.
(i) If CMS determines that the Biosimilar has not been licensed and marketed, subject to the circumstance in paragraph (h)(3) of this section, CMS includes the Reference Drug on the selected drug list for the initial price applicability year that is 2 years after the initial price applicability year for which the Reference Drug would have been included on the selected drug list if not for the successful Initial Delay Request.
(3) If one or more different biosimilars of the Reference Drug for the Biosimilar named in a successful Initial Delay Request (for purposes of paragraph (h)(1) of this section) or a successful Additional Delay Request (for purposes of paragraph (h)(2) of this section) are licensed and marketed, the Reference Drug is not included on the applicable selected drug list.
(i)
Rebate owed for failure of a biosimilar to be licensed and marketed.
(1) The Reference Manufacturer is required to pay the rebate specified in paragraph (i)(4) of this section for the 1 or 2 years that the Reference Manufacturer would have provided access to the MFP for the Reference Drug but for the Biosimilar Delay, if—
(i) CMS delayed the selection and negotiation of a Reference Drug for 1 year due to a successful Initial Delay Request or 2 years due to a successful Additional Delay Request;
(ii) CMS determined that the Biosimilar was not licensed and marketed, as described in paragraph (h) of this section; and
(iii) The Reference Manufacturer agrees to an MFP.
(2) The Reference Manufacturer must pay any rebates owed under paragraph (i) of this section in a time and manner to be specified by CMS.
(3) The rebates paid are deposited in either of the following:
(i) The Federal Supplementary Medical Insurance Trust Fund established under section 1841 of the Act, for drugs payable under Part B.
(ii) The Medicare Prescription Drug Account established under section 1860D-16 of the Act, within the Federal Supplementary Medical Insurance Trust Fund, for drugs covered under Part D.
(4) Except if the drug meets the circumstances in paragraph (i)(5) of this section, the rebate amount is calculated as follows:
(i) CMS applies the calculation under paragraphs (i)(4)(ii) through (iv) of this section, as applicable for all the previous initial price applicability years where the Reference Drug would have been on the selected drug list if not for the delay, but for the initial price applicability year for which an Additional Delay Request was granted, CMS adjusts the MFP by the annual percentage increase in the CPI-U for the 12-month period ending with July of the calendar year that is 2 years before the initial price applicability year for which the Additional Delay Request was granted.
(ii) In the case of a Reference Drug that is a drug covered under Part D—
(A) Seventy-five percent of the difference between the AMP as reported by the Primary Manufacturer of the selected drug under section 1927 of the Act, or if not reported under section 1927 of the Act, as submitted to CMS in accordance with § 429.200(b)(5), with respect to each of the calendar quarters of the price applicability period that would have applied but for the delay, and the MFP negotiated for the Reference Drug; and
(B) Multiplied by the number of units dispensed under Part D for the Reference Drug in each calendar quarter of the price applicability period that would have applied but for the delay.
(iii) In the case of a Reference Drug that is a drug payable under Part B—
(A) Eighty percent of the difference between the payment amount under section 1847A(b) of the Act, with respect to each of the calendar quarters of the price applicability period that would have applied but for the delay, and the MFP negotiated for the Reference Drug; and
(B) Multiplied by the number of units of the billing and payment code of the Reference Drug administered or furnished under Part B (excluding units that are packaged into the payment amount for an item or service and are not separately payable under Part B) for each calendar quarter of the price applicability period that would have applied but for the delay.
(iv) In the case of a Reference Drug that is a drug covered under Part D and payable under Part B, CMS calculates the rebate amount for Reference Drugs payable under Part B and covered under Part D by summing the rebate amount for the units payable under Part B as specified in paragraph (i)(4)(iii) of this section and the rebate amount for units covered under Part D as specified in paragraph (i)(4)(ii) of this section.
(5) If CMS determines that the Reference Drug transitioned from an extended-monopoly drug to a long-monopoly drug at the time of its inclusion on the selected drug list for an initial price applicability year (as set forth in § 429.110(h)(1)(ii) or (h)(2)), as applicable for the initial year delay—
(i) The rebate calculation as described in paragraph (i)(4) of this section will substitute the MFP negotiated for the
( printed page 36344)
Reference Drug with the amount specified in paragraph (i)(5)(ii) of this section.
(ii) The amount specified in this paragraph is an amount equal to 65 percent of the average non-FAMP for 2021 for the Reference Drug (or the first full year following market entry if there is no non-FAMP for 2021) increased by the percentage increase in the CPI-U from September 2021 (or December of such first full year following the market entry) to September of the year prior to the selected drug publication date for the initial price applicability year that would have applied but for the Initial Delay Request.
(6) If CMS determines that the Reference Drug transitioned from an extended-monopoly drug to a long-monopoly drug at the time of its inclusion on the selected drug list for an initial price applicability year (as set forth in § 429.110(h)(2)), as applicable for the second year delay—
(i) The rebate calculation as described in paragraph (i)(4) of this section will substitute the MFP negotiated for the Reference Drug with the amount specified at paragraph (i)(5)(ii) of this section that is further adjusted by the annual percentage increase in the CPI-U for the 12-month period ending with July of the calendar year that is 2 years before the initial price applicability year for which the Additional Delay Request was granted.
(a)
Identification.
With respect to each initial price applicability year beginning with initial price applicability year 2029, a qualifying single source drug, identified as set forth in § 429.125, is a negotiation-eligible drug if CMS determines that such qualifying single source drug is on the list of 50 Part D high spend drugs, as set forth in paragraph (a)(1) of this section, or on the list of 50 Part B high spend drugs or both, as set forth in paragraph (a)(2) of this section.
(1)
Part D high spend drugs.
CMS identifies Part D high spend drugs as follows:
(i) CMS removes a qualifying single source drug if it is currently a selected drug.
(ii) For each remaining qualifying single source drug identified in § 429.125, CMS calculates total expenditures under Part D using the methodology set forth at § 429.120(a).
(iii) CMS ranks the remaining qualifying single source drugs by total expenditures under Part D.
(iv) Subject to paragraph (b) of this section and using the ranked list identified in paragraph (a)(1)(iii) of this section, CMS identifies as Part D high spend drugs the 50 qualifying single source drugs that have the highest total expenditures under Part D.
(2)
Part B high spend drugs.
CMS identifies Part B high spend drugs as follows:
(i) CMS removes a qualifying single source drug if it is currently a selected drug.
(ii) For each remaining qualifying single source drug identified in § 429.125, CMS calculates total expenditures under Part B using the methodology set forth at § 429.120(b).
(iii) CMS ranks the remaining qualifying single source drugs by total expenditures under Part B.
(iv) Subject to paragraph (b) of this section and using the ranked list identified in paragraph (a)(2)(iii) of this section, CMS identifies as Part B high spend drugs the 50 qualifying single source drugs that have the highest total expenditures under Part B.
(b) If two or more qualifying single source drugs have the same total expenditures to the cent (and such total expenditures are the 50th highest among qualifying single source drugs under Part D (as specified in paragraph (a)(1)(iv) of this section) or Part B (as specified in paragraph (a)(2)(iv) of this section), CMS ranks those qualifying single source drugs based on which drug has the earlier approval or licensure date (as applicable) identified at § 429.125(c), until CMS has identified 50 high spend drugs for Part B and 50 high spend drugs for Part D, as applicable.
(a)
Total expenditures under Part D.
CMS calculates total expenditures under Part D for a given potential qualifying single source drug, qualifying single source drug, negotiation-eligible drug, or selected drug, as the sum of gross covered prescription drug costs for each PDE record for such drug that meets all of the following criteria:
(1) The date of service is during the total expenditures measurement period.
(2) The gross covered prescription drug cost on the PDE record is greater than zero dollars.
(3) The PDE record is considered final action.
(4) The drug coverage status code indicates the PDE record is for a drug covered under Part D.
(5) The compound code indicates the PDE record is not for a compounded drug.
(b)
Total expenditures under Part B.
CMS calculates total expenditures under Part B for a given potential qualifying single source drug, qualifying single source drug, negotiation-eligible drug, or selected drug, as the sum of the amounts determined under paragraphs (b)(1) and (b)(2) of this section:
(1) Subject to the methodology described in paragraph (b)(3) of this section, the sum of the total allowed charges for each Original Medicare Part B claim for such drug that meets all of the following criteria:
(i) The date of service is during the total expenditures measurement period.
(ii) The claim type code is associated with an Original Medicare Part B claim in an outpatient setting (including but not limited to clinics, Federally Qualified Health Centers, and ambulatory surgical centers), a professional services claim, or a durable medical equipment claim, as determined by CMS.
(iii) The total allowed charges for the claim line is greater than zero dollars.
(iv) The claim is considered final action.
(v) The claim is not billed as a compounded drug.
(vi) The claim is not for a drug or biological product that is bundled or packaged into the payment for another service. In instances where a claim for separate payment is submitted for a drug payable under Part B when such a claim is typically payable under Part B Original Medicare payment rules only as part of a bundled payment, such claim will be considered to be bundled or packaged into the payment for another service and will not be included in the total allowed charges calculation.
(2) Subject to the methodology described in paragraph (b)(3) of this section, the sum of the total allowed charges that would have been applicable under Original Medicare Part B for each MA encounter data record for Part B services for such drug that meets all of the following criteria:
(i) The date of service is during the total expenditures measurement period.
(ii) The claim type code is associated with an MA encounter record in an outpatient setting (including but not limited to clinics, Federally Qualified Health Centers, and ambulatory surgical centers), professional services, or durable medical equipment record, as determined by CMS.
(iii) The reported total number of units on the MA encounter data record line is greater than zero.
(iv) The encounter data record is considered final action.
(v) The encounter data record is not denied.
(vi) The encounter data record is not a chart review record.
( printed page 36345)
(vii) The encounter data record line is not for a supplemental benefit.
(viii) The encounter data record is not reported as a compounded drug.
(ix) The encounter data record is not for a drug or biological product that is bundled or packaged into the payment for another service under Part B Original Medicare. In instances where an encounter data record for separate payment is submitted for a drug payable under Part B when such a claim is typically payable under Part B Original Medicare payment rules only as part of a bundled payment, such claim will be considered to be bundled or packaged into the payment for another service and will not be included in the total allowed charges calculation.
(3) In cases where such drug is assigned to a HCPCS code with other products, CMS calculates the total expenditures under Part B by using ASP sales volume data to apportion Part B expenditures based on the ratio of reported sales volume of the drug compared to reported sales volume of all products assigned to the HCPCS code.
(a)
General.
Subject to the statutory exclusions codified in paragraph (e) of this section, with respect to each initial price applicability year beginning with initial price applicability year 2029, a qualifying single source drug is a drug covered under Part D, a drug payable under Part B, or both that meets the following criteria:
(1) For drug products, a qualifying single source drug is a drug—
(i) That is approved under section 505(c) of the FD&C Act and marketed in accordance with such approval;
(ii) For which, as of the selected drug publication date with respect to such initial price applicability year, at least 7 years have elapsed since the date of such approval, determined as set forth in paragraph (c) of this section; and
(iii) That is not the listed drug for any drug approved and marketed in an ANDA under section 505(j) of the FD&C Act, determined as set forth in paragraph (d) of this section.
(2) For biological products, a qualifying single source drug is a biological product—
(i) That is licensed under section 351(a) of the PHS Act and marketed in accordance with such licensure;
(ii) For which, as of the selected drug publication date with respect to such initial price applicability year, at least 11 years have elapsed since the date of such licensure, determined as set forth in paragraph (c) of this section; and
(iii) That is not the reference product for any biological product that is licensed and marketed under section 351(k) of the PHS Act, determined as set forth in paragraph (d) of this section.
(b)
Potential qualifying single source drugs.
In identifying qualifying single source drugs under paragraph (a) of this section, CMS first identifies potential qualifying single source drugs as follows:
(1) Subject to paragraph (b)(4) of this section, for drug products, all dosage forms and strengths of the drug with the same active moiety as identified using public sources (such as, but not limited to, RxNorm, OpenFDA, FDALabel, DailyMed, and FDA's Active Ingredient-Active Moiety Relationship/Basis of Strength file), and the same NDA holder, inclusive of all of the following:
(i) Products that are marketed under different NDAs.
(ii) Repackaged and relabeled products that are marketed under such NDA(s).
(iii) Authorized generic drugs, as defined in § 429.20, that are marketed under such NDA(s).
(iv) Multi-market approval (MMA) products imported under section 801(d)(1)(B) of the FD&C Act that are marketed under such NDA(s).
(2) Subject to paragraphs (b)(3) and (b)(4) of this section, for biological products, all dosage forms and strengths of the biological product with the same active ingredient as identified using public sources (such as, but not limited to, RxNorm, OpenFDA, FDALabel, DailyMed, and FDA's Active Ingredient-Active Moiety Relationship/Basis of Strength file), and the same BLA holder, inclusive of all of the following:
(i) Products that are marketed under different BLAs.
(ii) Repackaged and relabeled products that are marketed under such BLA(s).
(iii) Authorized generic drugs, as defined in § 429.20, that are marketed under such BLA(s).
(iv) MMA products imported under section 801(d)(1)(B) of the FD&C Act that are marketed under such BLA(s).
(3) Subject to paragraph (b)(4) of this section, for purposes of the identification of potential qualifying single source drugs under paragraph (b)(2) of this section for vaccines for infectious disease(s), CMS identifies all dosage forms and strengths of the drug with the same antigen component on such vaccine's labeling.
(4)
Fixed combination products.
Subject to paragraph (b)(4)(i) of this section, for purposes of the identification of potential qualifying single source drugs under paragraphs (b)(1) and (b)(2) of this section for a drug or biological product that is a fixed combination drug with two or more active moieties, active ingredients, or, for vaccines for infectious disease(s), antigen components, CMS identifies a potential qualifying single source drug using all dosage forms and strengths of the drug or biological product with the same distinct combination of active moieties, active ingredients, or antigen components.
(i)
New Formulations.
If CMS determines that a fixed combination drug with two or more active moieties, active ingredients, or, for a vaccine for infectious disease(s), antigen components shares one or more active moiety(ies), active ingredient(s), or antigen component(s) with another drug or biological product with the same NDA/BLA holder, and such products differ in active moiety(ies), active ingredient(s), or antigen component(s) due to the inclusion of an active moiety, active ingredient, or antigen component that creates a new formulation and enables an alternative route of administration for the co-administered active moiety(ies), active ingredient(s), or antigen component(s), CMS identifies a potential qualifying single source drug using all dosage forms and strengths of the drug or biological product with the shared active moiety(ies), active ingredient(s), or antigen component(s) and the same NDA/BLA holder.
(c)
Time since approval or licensure.
With respect to initial price applicability years beginning with initial price applicability year 2029, CMS determines if a potential qualifying single source drug, identified as set forth in paragraph (b) of this section, meets the time since approval or licensure criteria set forth in paragraphs (a)(1)(ii) and (a)(2)(ii) of this section, respectively, as follows:
(1)
Time since approval for drugs.
Subject to paragraph (c)(3) of this section, for a drug, at least 7 years must have elapsed between the date of approval of the potential qualifying single source drug and the selected drug publication date with respect to such initial price applicability year. To determine the FDA date of approval of a potential qualifying single source drug that is a drug, CMS uses the date of approval of the FDA application belonging to the NDA holder and containing the active moiety (or in the case of potential qualifying single source drugs identified under § 429.125(b)(4), the distinct combination of active moieties), or, if such drug has more than one FDA application, CMS uses the initial date of approval associated with the earliest-approved
( printed page 36346)
FDA application belonging to the NDA holder and containing the active moiety (or in the case of potential qualifying single source drugs identified under § 429.125(b)(4), the distinct combination of active moieties). For potential qualifying single source drugs identified under § 429.125(b)(4)(i), CMS uses the date of approval of the FDA application belonging to the NDA holder and containing the shared active moiety(ies) identified at § 429.125(b)(4)(i), or, if such drug has more than one FDA application, CMS uses the initial date of approval associated with the earliest-approved FDA application belonging to the NDA holder and containing such shared active moiety(ies).
(2)
Time since licensure for biological products.
Subject to paragraph (c)(3) of this section, for a biological product, at least 11 years must have elapsed between the date of licensure of the potential qualifying single source drug and the selected drug publication date with respect to such initial price applicability year. Subject to the exceptions set forth in paragraphs (c)(2)(i) and (c)(2)(ii) of this section, to determine the FDA date of licensure of a potential qualifying single source drug that is a biological product, CMS uses the date of licensure of the FDA application belonging to the BLA holder and containing the active ingredient (or in the case of potential qualifying single source drugs identified under § 429.125(b)(4), the distinct combination of active ingredients), or, if such biological product has more than one FDA application, CMS uses the initial date of licensure associated with the earliest-approved FDA application belonging to the BLA holder and containing the active ingredient (or in the case of potential qualifying single source drugs identified under § 429.125(b)(4), the distinct combination of active ingredients). Subject to the exceptions set forth in paragraphs (c)(2)(i) and (c)(2)(ii) of this section, for a potential qualifying single source drug identified under § 429.125(b)(4)(i), CMS uses the date of licensure of the FDA application belonging to the BLA holder and containing the shared active ingredient(s) identified at § 429.125(b)(4)(i), or, if such biological product has more than one FDA application, CMS uses the initial date of licensure associated with the earliest-approved FDA application belonging to the BLA holder and containing such shared active ingredient(s).
(i) For each unique potential qualifying single source drug that CMS identifies based on its antigen component(s) in accordance with paragraph (b)(3) of this section, CMS determines the date of licensure using the earliest date of licensure for any BLA or supplemental BLA for that unique potential qualifying single source drug for purposes of determining the FDA date of licensure of the potential qualifying single source drug for purposes of implementing paragraph (c)(2) of this section.
(ii) For biological products with an approved NDA that was deemed to be a BLA under section 351 of the PHS Act on March 23, 2020, in accordance with section 7002(e)(4)(A) of Biologics Price Competition and Innovation Act of 2009 (BPCI Act), and that are currently licensed biological products under section 351 of the PHS Act, CMS considers March 23, 2020 to be the date of licensure of the potential qualifying single source drug for purposes of implementing paragraph (c)(2) of this section. For a biological product with an approved application under section 505(c) of the FD&C Act that was deemed to be a BLA under section 7002(e)(4)(B) of the BPCI Act, as amended by the Further Consolidated Appropriations Act of 2020, CMS considers the approval date determined in accordance with section 7002(e)(4)(B) of the BPCI Act to be the date of licensure of the potential qualifying single source drug for purposes of implementing paragraph (c)(2) of this section.
(3)
Certain former orphan drugs.
Notwithstanding paragraphs (c)(1) and (c)(2) of this section to the contrary, for a drug or biological product that met or meets the criteria for the orphan drug exclusion at paragraph (e)(1) of this section as of the date of such drug or biological product's initial approval or licensure identified as set forth in paragraphs (c)(1) and (c)(2) of this section, CMS measures the 7- and 11-year periods described in paragraphs (c)(1) and (c)(2) of this section, respectively, starting from the first day after such initial date of approval or licensure that such drug or biological product does not, or did not, meet the criteria for the orphan drug exclusion. CMS identifies this day as the earlier of—
(i) The date on which the FDA approves such drug or biological product for an indication (irrespective of whether approval of such indication was or is withdrawn after its approval) for a disease or condition that is not a rare disease or condition for which the drug or biological product is designated under section 526 of the FD&C Act; or
(ii) The date on which an orphan drug designation is withdrawn, if that withdrawal results in the drug or biological product no longer qualifying for the orphan drug exclusion. For orphan drug designations withdrawn prior to or on August 12, 2013, for which the FDA Orphan Drug Product designation database does not include the date of such withdrawal, CMS uses August 12, 2013, as the date on which the orphan designation is withdrawn for purposes of identifying the first day after the drug or biological product's approval or licensure that such drug or biological product does not qualify for the orphan drug exclusion.
(d)
Bona Fide Marketing of an approved generic drug or licensed biosimilar.
A potential qualifying single source drug is not a qualifying single source drug, if CMS determines that—
(1) Using FDA reference sources including the Orange Book and Purple Book, at least one generic drug is approved under section 505(j) of the FD&C Act using any dosage form or strength of the potential qualifying single source drug as the listed drug or at least one biosimilar is licensed under section 351(k) of the PHS Act using any dosage form or strength of the potential qualifying single source drug as the reference product; and
(2) Such generic drug or biosimilar is subject to Bona Fide Marketing when CMS reviews such information at the point in time specified in § 429.130(c)(1).
(e)
Exclusions from qualifying single source drugs.
A potential qualifying single source drug identified in paragraph (b) of this section is not a qualifying single source drug if CMS determines that it qualifies for one or more of the following exclusions:
(1)
The orphan drug exclusion.
A potential qualifying single source drug qualifies for the orphan drug exclusion if, for all dosage forms and strengths of such potential qualifying single source drug, the following criteria are met:
(i) The potential qualifying single source drug is designated as a drug for one or more rare diseases or conditions under section 526 of the FD&C Act, determined by CMS as follows:
(A) CMS uses data sources such as the FDA Orphan Drug Product designation database; and
(B) CMS only considers orphan drug designations that have not been withdrawn or revoked.
(ii) The only indication(s) approved by FDA for the potential qualifying single source drug are for such designated rare disease(s) or condition(s), determined by CMS as follows:
(A) CMS uses information on FDA-approved indications from publicly available databases and documents (such as, but not limited to, FDALabel,
( printed page 36347)
FDA Online Label Repository,
Drugs@FDA,
and NLM DailyMed); and
(B) CMS only considers approvals of indications that FDA has not withdrawn.
(2)
The low-spend Medicare drug exclusion.
A potential qualifying single source drug qualifies for the low-spend Medicare drug exclusion if it has combined total expenditures under Part B and Part D, calculated as set forth in paragraph (e)(2)(i) of this section, less than the inflation-adjusted threshold, calculated as set forth in paragraph (e)(2)(ii) of this section.
(i) For a potential qualifying single source drug described in § 429.125(b), CMS calculates combined total expenditures under Part B and Part D as the sum of total expenditures under Part B (calculated as set forth in § 429.120(b)) plus total expenditures under Part D (calculated as set forth in § 429.120(a)).
(ii) Starting from the inflation-adjusted threshold for initial price applicability year 2028 equal to $212,907,518.30, the inflation-adjusted threshold for an initial price applicability year is equal to the inflation-adjusted threshold for the prior initial price applicability year, increased by the annual percentage increase in the CPI-U for the 12-month period ending on September 30 of the year prior to the year of the selected drug publication date with respect to a given initial price applicability year.
(3)
Plasma-derived product exclusion.
A potential qualifying single source drug qualifies for the plasma-derived product exclusion if it is a licensed biological product that is derived from human whole blood or plasma, as indicated on the approved product labeling or product information (such as, but not limited to, FDALabel and the FDA Online Label Repository).
(a)
General.
CMS considers whether an approved generic drug or licensed biosimilar is marketed according to this section based on CMS' evaluation of the following:
(1) Sales and utilization data for the generic drug or biosimilar from the following data sources for the following data periods:
(i) The 12 months of PDE data ending with the last full month of data available to CMS at the time CMS reviews such data consistent with paragraph (c) of this section;
(ii) The 12 months of AMP data ending with the last full month of data available to CMS at the time CMS reviews such data consistent with paragraph (c) of this section;
(iii) The 4 quarters of ASP data ending with the last full quarter of data available to CMS at the time CMS reviews such data consistent with paragraph (c) of this section; and
(iv) The 12 months or 4 quarters of any data source, if applicable to such data source, that is in addition to those data sources listed in paragraph (a)(1)(i) through (iii) of this section and may include, but is not limited to, Medicaid State Drug Utilization Data and data from nationally representative and commercially available databases, ending with the last full month or quarter of data available to CMS (as applicable to the specific data source) at the time CMS reviews such data consistent with paragraph (c) of this section.
(2) Information related to additional factors, including:
(i) Whether the generic drug or biosimilar is regularly and consistently available for purchase through the pharmaceutical supply chain; and
(ii) Whether any licenses or other agreements between a Primary Manufacturer and the manufacturer of the generic drug or biosimilar limit the availability or distribution of the generic drug or biosimilar.
(3) CMS may also use other available data and informational sources on market share and relative market competition of the generic drug or biosimilar.
(b)
Conduct holistic inquiry.
In its evaluation of the information described in paragraph (a) of this section, CMS conducts a holistic inquiry based on the totality of the circumstances.
(c)
Timing for CMS review of Bona Fide Marketing.
CMS reviews the information set forth in paragraph (a) of this section that is available at the following times:
(1)
Identification of potential qualifying single source drugs.
For any potential qualifying single source drug for initial price applicability year 2029 and any initial price applicability year thereafter, in January prior to each selected drug publication date.
(2)
Monitoring generic(s) or biosimilar(s) for drugs that were not considered qualifying single source drugs.
For any potential qualifying single source drugs that did not qualify as a qualifying single source drug for initial price applicability year 2029 or any initial price applicability year thereafter because CMS determined that a generic drug was approved using the potential qualifying single source drug as the listed drug or a biosimilar was licensed using the potential qualifying single source drug as the reference product, and such generic drug or biosimilar is subject to Bona Fide Marketing (consistent with paragraph (a) of this section), periodically during each calendar year, including at least annually in January.
(3)
Identification of selected drugs ineligible to be selected for renegotiation.
For identifying that a drug previously selected for negotiation is not eligible for renegotiation for initial price applicability year 2029 and any initial price applicability year thereafter, in January prior to each selected drug publication date.
(4)
Selected drugs until the negotiation period ends.
For drugs selected for negotiation for initial price applicability year 2029 and any initial price applicability year thereafter, monthly starting in March during the negotiation period for the initial price applicability year for which the drug was selected for negotiation and until November 1 of the year that begins 2 years prior to the initial price applicability year for which the drug was selected for negotiation.
(5)
Drugs selected for renegotiation until renegotiation ends.
For drugs selected for renegotiation (if any) for initial price applicability year 2029 and any initial price applicability year thereafter, monthly starting in March during the renegotiation period for the initial price applicability year for which the drug was selected for renegotiation and until November 1 of the year that begins 2 years prior to the initial price applicability year for which the drug was selected for renegotiation.
(6)
Selected drugs following the negotiation period.
For all drugs selected for initial price applicability year 2029 and any initial price applicability year thereafter after reaching an agreed-upon MFP (consistent with § 429.200(b)(4)), and, starting January 1, 2029, for any drugs selected for initial price applicability years 2026, 2027, and 2028, biannually in March and October, starting in October of the calendar year after CMS and the Primary Manufacturer reached an agreement on the MFP for the initial price applicability year for which the drug was selected originally for negotiation and until CMS determines that a selected drug meets the requirements at § 429.135(b) to cease being considered a selected drug.
(7)
Monitoring generic(s) or biosimilar(s) for selected drugs that are no longer subject to negotiation or that have ceased to be selected drugs.
For drugs selected for initial price applicability year 2029 and drugs selected for any initial price applicability year thereafter (consistent with § 429.200(b)(4)) for which CMS
( printed page 36348)
makes a determination under § 429.135(b), and, starting January 1, 2029, for any drugs CMS determines cease being considered a selected drug for initial price applicability years 2026, 2027, and 2028, periodically during each calendar year, including at least annually in January prior to each selected drug publication date.
(a)
General.
Each drug selected for negotiation for an initial price applicability year remains a selected drug with respect to such initial price applicability year and each subsequent year beginning before the first year that begins at least 9 months after the date as of which CMS determines that—
(1) Using FDA reference sources including the Orange Book and Purple Book, at least one generic drug is approved under section 505(j) of the FD&C Act using any dosage form or strength of the selected drug as the listed drug or at least one biosimilar is licensed under section 351(k) of the PHS Act using any dosage form or strength of the selected drug as the reference product; and:
(2) Such generic drug or biosimilar is subject to Bona Fide Marketing.
(b)
Timing.
For clarity, the following circumstances apply to such selected drug based on the date as of which CMS determines the conditions described in § 429.135(a) are met—
(1) If the date is during the period beginning on the selected drug publication date (as defined in § 429.20) and ending on November 1 of the year that begins 2 years prior to the initial price applicability year for which the drug is selected for negotiation, then—
(i) The selected drug ceases to be subject to the negotiation process under section 1194 of the Act, as set forth in subpart F of this part;
(ii) The selected drug remains a selected drug for such initial price applicability year only;
(iii) The selected drug will not be replaced with another selected drug for the initial price applicability year that the selected drug is selected; and
(iv) No MFP will be published for, or apply to, such drug.
(2) If the date is during a period beginning on the selected drug publication date (as defined in § 429.20) of the year that begins 2 years prior to the initial price applicability year for which the drug is selected for renegotiation and ending on November 1 of such year, then the selected drug ceases to be subject to the renegotiation process under section 1194 of the Act, as set forth in subpart G of this part.
(3) If the date is during the period beginning on November 2 of the year that begins 2 years prior to the initial price applicability year for which the drug is selected for negotiation and ending on March 31 of that initial price applicability year, then—
(i) The selected drug will cease to be a selected drug on January 1 of the year following the initial price applicability year for which such drug was selected for negotiation; and
(ii) The MFP will apply only for the initial price applicability year for which such drug was selected for negotiation.
(4) If the date is during the selected drug's price applicability period and after March 31 of the initial price applicability year for which the selected drug is selected for negotiation:
(i) The selected drug will cease to be a selected drug on January 1 of the year that begins at least 9 months after the date as of which CMS determines the conditions described in § 429.135(a) are met; and
(ii) The MFP will apply until such date that the selected drug ceases to be a selected drug as described in paragraph (b)(4)(i) of this section.
(c) A drug previously selected for negotiation is not eligible for renegotiation if CMS has made a determination described in section 429.135(a), prior to the next selected drug publication date for the initial price applicability year for which any drug could be selected for renegotiation.
(d)
Public notice.
If CMS makes a determination described in paragraph (a) of this section, CMS will announce such determination in a timeframe and manner specified by CMS.
(a)
General.
The Negotiation Program Agreement between the Primary Manufacturer, as defined in § 429.20, and CMS contains provisions regarding the requirements specified in paragraph (b) of this section, and may contain other provisions as determined by CMS, in accordance with section 1193 of the Act, as necessary for purposes of administering and monitoring compliance with the Negotiation Program.
(1) The deadline for the Primary Manufacturer of a selected drug to enter into a Negotiation Program Agreement with respect to the selected drug is 11:59 p.m. PST on February 28 following the selected drug publication date with respect to the initial price applicability year for which the selected drug was selected for negotiation.
(2) The negotiation period with respect to a selected drug will begin on the earlier of February 28 following the selected drug publication date with respect to the initial price applicability year for which the selected drug was selected for negotiation or the date that the Negotiation Program Agreement is fully executed.
(b)
Agreement requirements.
In executing a Negotiation Program Agreement with respect to a selected drug, a Primary Manufacturer agrees to the following:
(1) To comply with all applicable requirements and conditions set forth in sections 1191 through 1198 of the Act and all applicable guidance and regulations, including this part, implementing those provisions and any changes to the Act that affect the Negotiation Program.
(2) To negotiate to determine an MFP for the selected drug with CMS, during the negotiation period for the initial price applicability year for the selected drug, in accordance with section 1194 of the Act, including as described in subpart F of this part.
(3) As applicable, to renegotiate to determine an MFP for the drug selected for renegotiation with CMS, during the renegotiation period for the initial price applicability year for the drug selected for renegotiation, in accordance with section 1194 of the Act, including as described in subpart G of this part.
(4) To provide access to the MFP, including as renegotiated, with respect to the selected drug, during the selected drug's price applicability period, in accordance with section 1193(a)(3) of the Act, including as described in subpart B and subpart I of this part.
(5) To submit to CMS, in a form and manner specified by CMS, the information specified in sections 1191 to 1198 of the Act, the Negotiation Program Agreement, or this part, including, but not limited to, the information as specified at §§ 429.100(d), 429.405, 429.505(b), and, if applicable, 429.615(b)(1), in accordance with sections 1193(a)(4) and 1193(a)(5) of the Act, including as described in subparts B, E, F, and G of this part.
(6) To comply with requirements determined by CMS to be necessary for the purposes of administering the Negotiation Program and monitoring compliance with the Negotiation Program, in accordance with section 1193(a)(5) of the Act, including as described in subpart J of this part.
(c)
Execution of agreement.
The Negotiation Program Agreement must be signed, in a form and manner specified by CMS, by an authorized representative
( printed page 36349)
of the Primary Manufacturer and by CMS.
(d)
Agreement term.
The Negotiation Program Agreement takes effect on the date that the Negotiation Program Agreement is signed both by an authorized representative of the Primary Manufacturer and by CMS. The term of the Negotiation Program Agreement is from such effective date until the Negotiation Program Agreement is terminated in accordance with § 429.205(a), including as described in subpart C of this part.
(e)
Agreement to an MFP.
Upon agreeing to an MFP for a selected drug, CMS and the Primary Manufacturer of a selected drug will formalize the agreement to an MFP through an Addendum to the Negotiation Program Agreement that must be signed, in a form and manner specified by CMS, by an authorized representative of the Primary Manufacturer and by CMS.
(a)
Termination of Negotiation Program Agreement.
The Negotiation Program Agreement terminates effective as of the date of the earlier of—
(1) The first date that the selected drug covered by the Negotiation Program Agreement is no longer a selected drug consistent with CMS' determination in accordance with section 1192(c) of the Act as described at § 429.135.
(2) The date of termination established in paragraph (b)(5) of this section in connection with a Request to Terminate by the Primary Manufacturer submitted under paragraph (b) of this section.
(b)
Primary Manufacturer-requested Termination.
(1) A Primary Manufacturer that wishes to terminate a Negotiation Program Agreement may submit in writing a Request to Terminate, in a form and manner specified by CMS. Such Request to Terminate must include:
(A) A request for termination of the Primary Manufacturer's applicable program agreements, and
(B) An attestation that through the end of the price applicability period for the selected drug, the Primary Manufacturer shall not seek—
(i) To enter any subsequent applicable program agreement; or
(ii) Coverage for any of its drugs under the Manufacturer Discount Program.
(2) If CMS determines that the Primary Manufacturer's Request to Terminate complies with paragraph (b)(1), CMS—
(A) Confirms receipt of the Primary Manufacturer's notice; and
(B) Executes the actions with respect to termination of the Primary Manufacturer's applicable program agreements as described in paragraphs (b)(3), (b)(4), and (b)(5) of this section, if applicable.
(3) If CMS determines that the Primary Manufacturer's Request to Terminate complies with paragraph (b)(1), the Request to Terminate will constitute good cause for CMS to terminate the Primary Manufacturer's applicable program agreement(s) under the Manufacturer Discount Program in accordance with section 1860D-14C(b)(4)(B)(i) of the Act and § 423.2752(c)(1) of this chapter, and to expedite the date on which none of the drugs of the Primary Manufacturer are covered by an agreement under the Manufacturer Discount Program in accordance with § 423.2752(c)(1) of this chapter.
(4) If CMS determines that the Primary Manufacturer's Request to Terminate complies with paragraph (b)(1) of this section, the Request to Terminate will also constitute good cause for CMS to terminate the Primary Manufacturer's applicable program agreement(s) under the Medicaid Drug Rebate Program in accordance with section 1927(b)(4)(B)(i) of the Act and the Medicaid National Drug Rebate Agreement.
(5) Unless a Primary Manufacturer rescinds its Request to Terminate in accordance with paragraph (d) of this section, CMS terminates the Negotiation Program Agreement effective on the first date following the receipt of a Request to Terminate that CMS determines complies with paragraph (b)(1) of this section on which none of the drugs of the Primary Manufacturer are covered by an agreement under the Manufacturer Discount Program in accordance with § 423.2752(c)(1) of this chapter.
(c)
A Decision by the Primary Manufacturer not to execute a Negotiation Program Agreement.
(1) A Primary Manufacturer that does not wish to enter into a Negotiation Program Agreement in accordance with § 429.200 may submit in writing a Request to Terminate, in a form and manner specified by CMS, that meets the requirements described in paragraph (b)(1) of this section.
(2) In response to the Request to Terminate, CMS takes the steps described in paragraphs (b)(2) through (b)(4) of this section, as applicable.
(d)
Request by a Primary Manufacturer to Rescind the Request to Terminate.
If a Primary Manufacturer wishes to rescind the Request to Terminate it submitted under paragraphs (b) or (c) of this section, it must file a written request for a hearing, in a form and manner specified by CMS.
(1) Upon such a request from the Primary Manufacturer, CMS provides a hearing concerning termination of the Primary Manufacturer's applicable program agreements, in accordance with sections 1860D-14C(b)(4)(B)(i) and 1927(b)(4)(B)(i) of the Act, as applicable.
(2) Such hearing will be held prior to the effective date of termination of the applicable program agreements with sufficient time for such effective date to be repealed.
(3) Such hearing will be held solely on the papers.
(4) The only question to be decided in the hearing is whether the Primary Manufacturer has asked to rescind its Request to Terminate prior to the effective date of termination of the applicable program agreements.
(e)
Effect of termination.
Notwithstanding termination of the Negotiation Program Agreement for a selected drug in accordance with either paragraph (a)(1) or (a)(2) of this section, the Primary Manufacturer is responsible for making the MFP for the selected drug available, in accordance with subpart I of this part, with respect to dispenses, administrations, and furnishings of the selected drug prior to the effective date of termination under paragraph (a) of this section. Confidentiality, record retention and data requirements and requirements for Primary Manufacturer participation in audit and other Negotiation Program oversight activities continue to apply.
(f)
Reentry into the Negotiation Program.
A Primary Manufacturer that has terminated the Negotiation Program Agreement for a selected drug in accordance with paragraph (b) of this section and later seeks to re-enter any applicable program agreement or obtain coverage for any of its drugs under the Manufacturer Discount Program during the selected drug's price applicability period would be deemed to have provided an invalid attestation, described in section § 429.205(b)(1)(B), and the Negotiation Program Agreement would once again become operative as of the date of re-entry into the applicable program agreement or coverage for any of its drugs under the Manufacturer Discount Program.
Other provisions of the Negotiation Program Agreement.
(a)
General.
If any provision of the Negotiation Program Agreement is found to be invalid by a court of law with competent jurisdiction, the Negotiation Program Agreement will be construed in all respects as if any
( printed page 36350)
invalid or unenforceable provision(s) were eliminated, and without any effect on any other provision.
(b)
Update to the Negotiation Program Agreement.
CMS retains the authority to amend the Negotiation Program Agreement to reflect changes in law, regulation, or guidance as applicable.
(c)
Transfer of Negotiation Program Agreement.
If, after entering in a Negotiation Program Agreement with CMS, the Primary Manufacturer of a selected drug transfers ownership of one or more NDAs or BLAs, as applicable, of a selected drug to another entity, the Primary Manufacturer remains responsible for all requirements of the Negotiation Program Agreement associated with the transferred NDA(s) or BLA(s), including the requirement to provide access to the MFP in accordance with subpart I, unless and until the Primary Manufacturer transfers all the NDAs or BLAs of the selected drug that it holds to an entity and such acquiring entity assumes responsibility as the new Primary Manufacturer in accordance with this paragraph (c).
(1) To transfer responsibility for all requirements of the Negotiation Program Agreement to an acquiring entity, the transferring Primary Manufacturer must—
(i) Transfer all NDA(s) or BLA(s) of the selected drug that it holds to the acquiring entity;
(ii) Provide CMS with documentation of the intended transfer of responsibility for all requirements of the Negotiation Program Agreement to the acquiring entity, in the form of a novation, at least 30 calendar days before the intended effective date of any such transfer for CMS review and approval. Such novation of the transferring Primary Manufacturer's Negotiation Program Agreement must be signed by the transferring Primary Manufacturer and the acquiring entity and must include, at minimum, the legal name of the acquiring entity, the effective dates of the transfer of ownership of all transferred NDAs or BLAs of the selected drug that the transferring Primary Manufacturer holds and of the transfer of responsibility for all requirements of the Negotiation Program Agreement, a list of all transferring NDAs or BLAs of the selected drug, and agreement that the acquiring entity assumes all obligations and liabilities under the transferring Primary Manufacturer's Negotiation Program Agreement as the successor in interest.
(2) If the transferring Primary Manufacturer submits a novation agreement that meets the requirements of paragraph (c)(1)(ii) of this section and is approved and signed by CMS, the acquiring entity becomes the successor in interest to the transferring Primary Manufacturer's Negotiation Program Agreement and the Primary Manufacturer of the applicable selected drug as of the novation's effective date of the transfer of responsibility for all requirements of the Negotiation Program Agreement.
(3) The transferring Primary Manufacturer remains responsible for any outstanding Negotiation Program rebate liabilities related to the Biosimilar Delay, under section 1192(f) of the Act, unless and until such liabilities are transferred to the acquiring entity as the new Primary Manufacturer, in accordance with paragraph (c) of this section.
(a)
Confidentiality of proprietary information.
Information deemed proprietary under this section will only be used by CMS or disclosed to and used by the Comptroller General of the United States for purposes of carrying out the Negotiation Program.
(b)
Confidentiality policy.
CMS would deem proprietary information, including trade secrets and confidential commercial or financial information, as confidential information exempt from disclosure if the information meets the requirements set forth under Exemption 3 or Exemption 4 of the Freedom of Information Act (5 U.S.C. 552(b)(3), (4)).
(1) CMS would not disclose protected health information or personally identifiable information except in accordance with applicable laws, where received by CMS as set forth in §§ 429.505 and 429.615 or received by CMS in engagements with interested parties specified in §§ 429.515 and 429.620(e).
(c)
Primary Manufacturer data.
The following data when submitted by the Primary Manufacturer as set forth at §§ 429.405 and 429.505(b), and 429.615(b)(1) would be deemed proprietary information by CMS, unless the information is publicly available:
(1) Non-FAMP and associated non-FAMP data collection;
(2) Research and development costs of the Primary Manufacturer for the selected drug;
(3) Current unit costs of production and distribution of the selected drug;
(4) Data on pending patent applications for the selected drug; and
(5) Market data and revenue and sales volume for the selected drug in the United States.
(6) A common Technical File/Drug Master File/“drug dossier” if submitted with the submission specified at § 429.505(d)(3) or § 429.615(b)(1)(i) (if applicable).
(d)
Publication.
Any information deemed proprietary by CMS or covered by the confidentiality policy, in accordance with paragraphs (a) and (b) of this section, must be redacted from any publications related to the Negotiation Program, including the publication of the MFP, as described in § 429.705.
Subpart E—Establishment of a Single MFP and Determination of the Ceiling
Establishment of a single MFP for negotiation and renegotiation purposes.
(a)
Establishment of a single MFP for negotiation and renegotiation purposes.
CMS identifies, for purposes of determining offers and counteroffers for use at each step of the negotiation process, as set forth in §§ 429.510 through 429.535, and for use at each step of the renegotiation process, as set forth in § 429.620, a single price for a selected drug, including for a selected drug with multiple dosage forms and strengths.
(1) CMS bases the single price on the cost of the selected drug per 30-day equivalent supply for all formulations weighted across dosage forms and strengths, as set forth in § 429.415.
(a) In accordance with the requirements of the Negotiation Program Agreement as set forth in § 429.200, the Primary Manufacturer must submit the following information to CMS in a form and manner specified by CMS by 11:59 p.m. PST on March 1 of the year of the selected drug publication date with respect to the initial price applicability year for which the selected drug was selected for negotiation, inclusive of the NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer:
(1) Non-FAMP, unit type, and total unit volume for each NDC-11 of the selected drug (as determined in § 429.100(d)) for the four quarters of calendar year 2021 in which the selected drug was sold and non-FAMP data was reported (or, in the case that there is not an average non-FAMP available for such selected drug for calendar year 2021 (when non-FAMP data has not been reported for at least one NDC-11 of the selected drug for at least one quarter in calendar year 2021)), the Primary Manufacturer is required to report the non-FAMP, unit type, and total unit volume for the four quarters of the first full calendar year
( printed page 36351)
following the market entry for such drug); and
(2) Non-FAMP, unit type, and total unit volume for each NDC-11 of the selected drug (as determined in § 429.100(d)) for the four quarters of the calendar year prior to the selected drug publication date with respect to the initial price applicability year for which the selected drug was selected for negotiation.
(b) In accordance with the requirements of the Negotiation Program Agreement as set forth in § 429.200, if the information in paragraph (a) of this section is restated due to requirements of 38 U.S.C. 8126 and implementing regulations and guidance issued by the Department of Veterans Affairs, the Primary Manufacturer is required to update the information submitted in accordance with paragraph (a) of this section in a form and manner specified by CMS.
(a)
Limitations on offer amount.
In negotiating the MFP of selected drugs with respect to their initial price applicability year, beginning with initial price applicability year 2029, and in accordance with section 1194(b)(2)(F)(i) of the Act, CMS will not make an offer (or agree to a counteroffer) for an MFP that exceeds the ceiling specified in section 1194(c) of the Act, as determined under this section, subject to § 429.440(b)(4), or is less than the Temporary Floor for Small Biotech Drugs specified in section 1194(d) of the Act, as determined under § 429.440(b)(3), if applicable.
(b)
Determination of the ceiling for the MFP.
With respect to each initial price applicability year beginning with initial price applicability year 2029, the MFP negotiated under the process set forth in §§ 429.510 through 429.535 for a selected drug with respect to the first initial price applicability year of the price applicability period with respect to such drug must not exceed the lower of the amount specified in paragraph (b)(1) or (b)(2) of this section. The ceiling for renegotiation is set forth in § 429.620(b).
(1) The amount determined under this paragraph equals one of the following amounts, as applicable to the selected drug:
(i) For a selected drug that is covered under Part D but is not payable under Part B, the sum of the plan-specific enrollment weighted amounts, as set forth in § 429.420.
(ii) For a selected drug that is payable under Part B and paid according to section 1847A(b)(4) of the Act, but is not covered under Part D, the payment amount under section 1847A(b)(4) of the Act, as set forth in § 429.425;
(iii) For a selected drug that is payable under Part B and paid according to section 1847A(b)(4) of the Act, and covered under Part D, an amount equal to the combined Part B and Part D amount, as set forth in § 429.430.
(iv) For a selected drug that is payable under Part B but is not paid according to section 1847A(b)(4) of the Act, and is covered under Part D, the sum of the plan-specific enrollment weighted amounts, as set forth in § 429.420.
(v) For a selected drug that is payable under Part B but is not paid according to section 1847A(b)(4) of the Act, and is not covered under Part D, there is no amount under paragraph (b)(1) of this section.
(2) The amount determined under this paragraph is equal to the applicable percent, as applicable to the selected drug, of the lower of—
(i) The average non-FAMP for calendar year 2021, or, as applicable, the first full year after 2021 for which data is available, adjusted by inflation, as set forth in § 429.435(a)(1); or
(ii) The average non-FAMP for the year preceding the selected drug publication with respect to the first initial price applicability year of the price applicability period for which the drug is being negotiated, or, as applicable, the most recent year prior for which data is available, as set forth in § 429.435(a)(2).
(3) CMS calculates a single amount, a 30-day equivalent supply as described in § 429.415, across all dosage forms and strengths of the selected drug for the amounts set forth in paragraphs (b)(1) and (b)(2) of this section to determine which amount is lowest and will serve as the ceiling for the MFP.
(c)
Suggestion of Error.
A Primary Manufacturer that believes in good faith that CMS has made an error in the calculations specified in paragraph (b) of this section may submit a Suggestion of Error, as set forth in § 429.445(c) and in a form and manner specified by CMS, as set forth in § 429.445(d).
(a)
Determination of the 30-day equivalent supply for a selected drug.
CMS calculates the 30-day equivalent supply across all dosage forms and strengths of the selected drug using the following methodologies, as applicable:
(1)
30-day equivalent supply for a selected drug covered under Part D.
For a selected drug that is covered under Part D, CMS uses the methodology in accordance with § 423.104(d)(2)(iv)(A)(2).
(2)
30-day equivalent supply for a selected drug covered under Part B.
For selected drugs that are payable under Part B, CMS determines the 30-day equivalent supply using the following methodology:
(i) For Part B data for the selected drug's HCPCS code(s) that is/are for the year prior to the year of the selected drug publication date with respect to the initial price applicability year (for which the selected drug is selected for negotiation), CMS identifies any subsequent instances of Part B data or any PDE record that:
(A) Is associated with the same beneficiary; and
(B) Is for a drug or biological product with the same active moiety/active ingredient, identified as set forth at § 429.125(b), as the selected drug.
(ii) CMS calculates a “days between services” amount by counting the days between the date of service on the first instance of Part B data and the date of service on the immediately subsequent instance of Part B data or PDE record.
(A) Notwithstanding paragraph (a)(2)(ii) of this section, if the beneficiary does not have a subsequent instance of Part B data or PDE record with the same active moiety/active ingredient, identified as set forth at § 429.125(b), as the selected drug, CMS assigns a “days between services” amount equal to the median “days between services” amount for all other instances of Part B data or PDE records for that selected drug associated with that beneficiary during the applicable claims period.
(B) Notwithstanding paragraph (a)(2)(ii)(A) of this section, if there are no other instances of Part B data or PDE records for that selected drug associated with that beneficiary during the year set forth in paragraph (a)(2)(i) of this section, CMS does not assign a “days between services” amount, and the claim will not be included in the calculations.
(iii) For each instance of Part B data or PDE record, the 30-day equivalent supply for the HCPCS code is equal to the “days between services” (as determined in paragraph (a)(2)(ii) of this section) divided by 30.
(iv) CMS allocates a portion of the HCPCS code's 30-day equivalent supply to each NDC within the HCPCS code for calculations that require a 30-day equivalent supply at the NDC level and will proceed in accordance with paragraph (v) of this section.
(v) To determine the total Part B 30-day equivalent supply at the NDC level, CMS takes the following steps:
(A) As set forth in § 429.100(f), use NDC-11s from the list of NDC-11s of
( printed page 36352)
the selected drug as set forth in paragraph § 429.100(c) to determine which NDC-11s of the selected drug are included in the calculations for the calendar year as set forth in § 429.425(a) that meet the following criteria:
(1)
The NDC-11 is assigned to the Primary Manufacturer or manufactured, marketed, controlled, or sold by a Secondary Manufacturer(s);
(2)
The NDC-11 does not represent a sample package;
(3)
The NDC-11 is assigned to a HCPCS code;
(4)
The NDC-11 does not represent a self-administered drug; and
(5)
CMS observes any instances of Part B data for the HCPCS code to which the NDC-11 is assigned during the calendar year as set forth in paragraph (a)(2)(i) of this section.
(B) Determine the total billing units sold for each NDC-11 assigned to the HCPCS code (including NDC-11s that do not belong to the selected drug, if applicable) for each quarter of the calendar year set for in paragraph (a)(2)(1) of this section, by multiplying the number of units reported by a manufacturer in ASP data submissions at the NDC-11 package level by the number of billing units per NDC-11 reporting unit.
(C) For all NDC-11s assigned to the HCPCS code that is associated with the selected drug, sum the total billing units sold for such NDC-11s across all calendar quarters to calculate an annual total billing units sold for all NDC-11s assigned to the HCPCS code.
(D) For each NDC-11 assigned to the HCPCS code that is associated with the selected drug, divide the annual total billing units sold for that NDC-11 by the annual total billing units sold for all applicable NDC-11s of the same HCPCS code.
(E) For each NDC-11 assigned to the HCPCS code that is associated with the selected drug, multiply the 30-day equivalent supply for the HCPCS code (as determined in paragraph (a)(2)(iii) of this section) by the quotient calculated in paragraph (a)(2)(v)(D) of this section, to yield the total 30-day equivalent supply for the NDC-11.
(1)
Notwithstanding paragraph (a)(2)(ii)(B) of this section, CMS plans to assign a value of “12” for the 30-day equivalent supply for drugs typically administered one time (for example, some vaccines and cancer therapies).
(b)
Suggestion of Error.
A Primary Manufacturer that believes in good faith that CMS has made an error in the calculations specified in paragraph (a) of this section may submit a Suggestion of Error, as set forth in § 429.445(c) and in a form and manner specified by CMS, as set forth in § 429.445(d).
Determination of the sum of the plan-specific enrollment weighted amounts.
(a)
Data used to calculate the sum of the plan-specific enrollment weighted amount for selected drugs as set forth in § 429.410(b)(1)(i) and (iv).
CMS uses plan sponsors' reported Part D PDE data and direct and indirect remuneration data for all Part D plans found in such PDE data that meet the inclusion and exclusion criteria as set forth in § 429.120(a)(2) through (5) for the year as set forth in paragraph (b)(1) of this section.
(1) As set forth in § 429.100(f), CMS uses NDC-11s from the list of NDC-11s of the selected drug as set forth in paragraph § 429.100(c) to determine which NDC-11s of the selected drug are included in the ceiling calculations for the calendar year as set forth in (b)(1) of this section that meet the following criteria:
(i) The NDC-11 is assigned to the Primary Manufacturer or manufactured, marketed, controlled, or sold by a Secondary Manufacturer(s);
(ii) The NDC-11 does not represent a sample package;
(iii) CMS observes any PDE days' supply, PDE quantity dispensed, and PDE gross expenditures during the calendar year as set forth in (b)(1) of this section; and
(iv) CMS observes any associated direct and indirect remuneration amounts for the NDC-11 during the calendar year as set forth in (b)(1) of this section.
(v) The PDE record meets the inclusion and exclusion criteria as set forth in § 429.120(a)(2) through (a)(5).
(b)
Determination of the sum of the plan-specific enrollment weighted amounts.
Using the 30-day equivalent supply methodology set forth at 42 CFR 423.104(d)(2)(iv)(A)(2), CMS calculates the sum of the plan-specific enrollment weighted amount for selected drugs as set forth in § 429.410(b)(1)(i) and (iv) to determine the sum of the plan-specific enrollment weighted amounts for across all NDCs of the selected drug by conducting the following steps:
(1) For each Part D plan, CMS identifies the PDE data for the selected drug's NDC-11s, as identified using the criteria set forth in paragraph (a)(1) of this section, for the most recent year for which all data are available.
(2) For each Part D plan and each NDC-11, CMS separately sums the negotiated price amounts (as defined in 42 CFR 423.100), the ERPOSA (as defined in § 429.20), and units dispensed.
(3) For each Part D plan and each NDC-11, CMS sums the total direct and indirect remuneration amounts found in the Detailed Direct and Indirect Remuneration Report for the year set forth in paragraph (b)(1) of this section and subtracts the total ERPOSA calculated in paragraph (b)(2) of this section.
(4) For each Part D plan and each NDC-11, CMS subtracts the total direct and indirect remuneration amounts minus ERPOSA amount calculated in paragraph (b)(3) of this section, from the total negotiated price amounts calculated in paragraph (b)(2) of this section, and then divide by the total units dispensed also determined in paragraph (b)(2) of this section.
(5) Separately, CMS identifies the total number of individuals enrolled in all Part D plans in December for the year set forth in paragraph (b)(1) of this section and the total number of individuals enrolled in each Part D plan in that same month. The Part D plans included in the calculations of this step will be restricted to Part D plans with at least one PDE record for that NDC-11 identified in paragraph (b)(1) of this section.
(6) For each Part D plan and each NDC-11, CMS divides the total number of Part D beneficiaries enrolled in the Part D plan as identified in paragraph (b)(5) of this section by the total number of individuals enrolled in all Part D plans also as identified in paragraph (b)(5) of this section, and multiplies this quotient by the price per unit, net of all price concessions received by such plan or pharmacy benefit manager on behalf of such Part D plan (as calculated in paragraph (b)(4) of this section) to arrive at the plan-specific enrollment weighted amount.
(7) For each NDC-11, CMS sums the amounts calculated in paragraph (b)(6) of this section across all Part D plans to calculate the sum of the plan-specific enrollment weighted amounts.
(8) For each NDC-11, CMS multiplies the sum of the plan-specific enrollment weighted amounts (as calculated in paragraph (b)(7) of this section), which are a per-unit price, by the NDC-11 average number of units per 30-day equivalent supply calculated from PDE data for the year set forth in paragraph (b)(1) of this section, (that is, quotient of the total quantity dispensed and the total 30-day equivalent supply as described in paragraph (b)(8)(i)) to yield the price of a 30-day equivalent supply. This calculation results in the sum of the plan-specific enrollment weighted amounts for each NDC-11 identified based on the criteria as set forth in (a)(1) of this section.
( printed page 36353)
(i) For each NDC-11, CMS calculates the quotient of total dispensed and total 30-day equivalent supply to calculate the NDC-11 average number of units per 30-day equivalent supply.
(9) For each NDC-11, CMS divides the total 30-day equivalent supply for that NDC-11 by the total 30-day equivalent supply across all NDC-11s of the selected drug, both calculated from PDE data from the year set forth in paragraph (b)(1) of this section and multiplies this quotient by the sum of the plan-specific enrollment weighted amounts for a 30-day equivalent supply as calculated in paragraph (b)(8) of this section.
(10) CMS sums amounts calculated in paragraph (b)(9) of this section, across all NDC-11s of the selected drug to generate the sum of the plan-specific enrollment weighted amounts for the selected drug for a 30-day equivalent supply.
(c)
Suggestion of Error.
A Primary Manufacturer that believes in good faith that CMS has made an error in the calculations specified in paragraph (b) of this section may submit a Suggestion of Error, as set forth in § 429.445(c) and in a form and manner specified by CMS, as set forth in § 429.445(d).
Determination of the payment amount under section 1847A(b)(4) of the Act.
(a)
Determination of the payment amount under section 1847A(b)(4) of the Act.
CMS calculates the payment amount under section 1847A(b)(4) of the Act, which is 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation, for a 30-day equivalent supply across all dosage forms and strengths of a selected drug.
(1) As set forth in § 429.100(f), CMS uses NDC-11s from the list of NDC-11s of the selected drug as set forth in paragraph § 429.100(c) to determine which NDC-11s of the selected drug are included in the ceiling calculations for the calendar year as set forth in paragraph (a) of this section that meet the following criteria:
(i) The NDC-11 is assigned to the Primary Manufacturer or manufactured, marketed, controlled, or sold by a Secondary Manufacturer(s);
(ii) The NDC-11 does not represent a sample package;
(iii) The NDC-11 is assigned to a HCPCS code;
(iv) The NDC-11 does not represent a self-administered drug; and
(v) CMS observes any instances of Part B data for the HCPCS code to which the NDC-11 is assigned during the calendar year as set forth in § 429.425(a).
(vi) The Part B data meets the inclusion and exclusion criteria as set forth in § 429.120(b)(1)(ii) through (vi) and (b)(2)(ii) through (ix).
(2)
Calculating the payment amount under section 1847A(b)(4) of the Act.
CMS calculates the payment amount under section 1847A(b)(4) of the Act for each HCPCS code to which any NDC-11 identified in paragraph (a)(1) are assigned, and then assigns such payment amount to each NDC-11 of the selected drug within such HCPCS code.
(i) CMS calculates separately, the annual ASP and WAC amounts for the HCPCS code, by converting quarterly ASP and WAC data for the year set forth in paragraph (a) of this section, for each NDC-11 of a selected drug as set forth in § 429.100(c) that is associated with a HCPCS code, to an annual calendar year ASP and WAC amount for each HCPCS code by taking an average of the reported ASP and WAC amounts for the NDC-11s associated with the HCPCS code across all four quarters of such calendar year, weighted by the total number of billing units in the Part B data for that HCPCS code each quarter.
(A) If the total number of billing units in the Part B data for the HCPCS code are zero for a given quarter, CMS assigns that quarter the lowest positive total units from among the other quarters in the same calendar year for that HCPCS code.
(B) For each of the separate ASP and WAC calculations, if the reported price is negative, zero, or missing for all applicable NDC-11s assigned to the HCPCS code in a given quarter for the calendar year as set forth in paragraph (a) of this section, CMS excludes that quarter from the applicable calculation.
(C) If the WAC reported to the ASP portal is negative, zero, missing for all applicable NDC-11s assigned to the HCPCS code for all four quarters of the calendar year as set forth in § 429.425(a) but the reported ASP is positive for at least one of the NDC-11s assigned to the HCPCS code, CMS uses the WAC reported by the Primary Manufacturer of the selected drug to CMS under section 1194(e)(1) of the Act as described in § 429.505(a).
(ii) CMS determines the lesser of the annual ASP and the annual WAC as determined under paragraph (a)(2)(i) of this section to yield the payment amount under section 1847A(b)(4) of the Act for the associated HCPCS code. This amount will apply to all applicable NDC-11s assigned to the associated HCPCS code.
(iii) Notwithstanding paragraphs (a)(2)(i) and (ii) of this section, if ASP, WAC reported to the ASP payer portal, and WAC reported by the Primary Manufacturer pursuant to section 1194(e)(1) of the Act as described in § 429.505(b)(2) are negative, zero, or missing for all applicable NDC-11s assigned to the HCPCS code for all four quarters of the calendar year as set forth in paragraph (a) of this section, CMS takes the average of the published payment limits in the ASP pricing file (or in the OPPS Addendum B file, if it is not available in the ASP pricing file) for the HCPCS code across all four quarters, weighted by the total number of billing units in the Part B data for that HCPCS code for the four quarters as described in paragraph (a)(2)(i) of this section (including the adjustments made when billing units zero) to calculate the payment amount under section 1847A(b)(4) of the Act.
(3)
Allocating HCPCS code-level utilization from Part B data for Part B services.
CMS allocates HCPCS code-level utilization from Part B data across each NDC-11 of the selected drug, identified in paragraph (a)(1) of this section, assigned to such HCPCS code.
(i) CMS allocates HCPCS code-level utilization from Part B data using the proportion of ASP units reported by manufacturers to CMS for each NDC-11 that is assigned to the HCPCS code.
(ii) CMS converts the total billing units in Part B data for the HCPCS code that includes the selected drug to determine the units that are comparable to NCPDP units that are used in PDE records of the total Part B billing units for the NDC-11 of the selected drug using the following steps:
(A) For each NDC-11 assigned to a HCPCS code to which an NDC-11 of the selected drug is assigned for each quarter (including NDC-11s that do not belong to the selected drug, if applicable), calculates the total number of billing units sold for each NDC-11 by multiplying the number of units reported by a manufacturer at the NDC-11 package level by the number of billing units per NDC-11 reporting unit.
(B) Divides the billing units sold for a NDC-11, as calculated in paragraph (a)(3)(ii)(A) of this section, by the total billing units sold for all NDC-11s within the same HCPCS code.
(C) Multiplies the total billing units from Part B data for the HCPCS code by the quotient calculated in paragraph (a)(3)(ii)(B) of this section, to determine the total Part B billing units for each NDC-11 of the selected drug.
(D) Notwithstanding paragraphs (a)(3)(ii)(A) through (C) of this section, if there are no units as measured on PDE records, the Part B unit type is not converted to a PDE unit type.
( printed page 36354)
(4)
Calculating the payment amount under section 1847A(b)(4) of the Act for a 30-day equivalent supply of the selected drug.
CMS calculates the payment amount under section 1847A(b)(4) of the Act for a 30-day equivalent supply of each NDC-11 identified based on the criteria as set forth in paragraph (a)(1) of this section.
(i) For each selected drug, CMS identifies the NDC-11s and the unique HCPCS code(s) associated with the selected drug using the criteria set forth in paragraph (a)(1) of this section.
(A) CMS identifies the annual calendar year set forth in paragraph (a) of this section, for the payment amount under section 1847A(b)(4) of the Act for each HCPCS code and assigns it to each NDC-11 within that HCPCS code.
(ii) CMS sums the converted Part B units from Part B data attributed to each NDC-11 (as calculated in paragraph (a)(3) of this section), across all four quarters of the calendar year as set forth in paragraph (a) of this section to calculate the annual Part B units of that NDC-11.
(iii) CMS calculates the total Part B 30-day equivalent supply in Part B data attributed to that NDC-11 of calendar year as set forth in § 429.425(a).
(iv) CMS calculates the quotient of the annual Part B units in Part B data for each NDC-11 of the selected drug (as calculated in paragraph (a)(4)(ii) of this section), and the total Part B 30-day equivalent supply in Part B data (as calculated in paragraph (a)(4)(iii) of this section) which is the average number of units per 30-day equivalent supply.
(v) CMS multiplies the payment amount under section 1847A(b)(4) of the Act (as determined in paragraph (a)(4)(i) of this section), by the average number of units per 30-day equivalent supply (as calculated in paragraph (a)(4)(iv) of this section), to yield the payment amount under section 1847A(b)(4) of the Act per 30-day equivalent supply for each NDC-11 of the selected drug.
(vi) CMS divides the total 30-day equivalent supply for each NDC-11 (as calculated in paragraph (a)(4)(iii) of this section) by the total 30-day equivalent supply across all NDC-11s of the selected drug and multiplies this quotient by the payment amount under section 1847A(b)(4) of the Act per 30-day equivalent supply for that NDC-11 (as calculated in paragraph (a)(4)(v) of this section).
(vii) CMS sums the amounts calculated in paragraph (a)(4)(v)(B) of this section, across all NDC-11s of the selected drug to generate the payment amount under section 1847A(b)(4) of the Act for the selected drug for a 30-day equivalent supply.
(b) CMS does not apply the sequestration payment adjustment (as defined in § 429.20 of this chapter) to the payment amount under section 1847A(b)(4) of the Act as part of this methodology to calculate the payment amount under section 1847A(b)(4) of the Act.
(c)
Suggestion of Error.
A Primary Manufacturer that believes in good faith that CMS has made an error in the calculations specified in paragraphs (a)(2) through (a)(4) of this section may submit a Suggestion of Error, as set forth in § 429.445(c) and in a form and manner specified by CMS, as set forth in § 429.445(d).
Determination of the combined Part B and Part D amount.
(a)
Determination of the combined Part B and Part D amount.
CMS calculates a single amount referred to as the combined Part B and Part D amount for a selected drug as set forth in § 429.410(b)(1)(iii) using the following methodology.
(1) CMS calculates the sum of the plan-specific enrollment weighted amounts, as set forth in § 429.420, and the payment amount under section 1847A(b)(4) of the Act, as set forth in § 429.425, for all dosage forms and strengths of a selected drug that is payable under Part B and covered under Part D as a 30-day equivalent supply by:
(i) Separately calculating the sum of the plan-specific enrollment weighted amounts, as set forth in paragraph § 429.420, and the payment amount under section 1847A(b)(4) of the Act, as set forth in paragraph § 429.425, using the following steps:
(A) If an NDC-11 is only present in PDE data, CMS calculates only the sum of the plan-specific enrollment weighted amounts per 30-day equivalent supply for that NDC-11 as set forth in paragraph § 429.420.
(B) If an NDC-11 is only associated with a Part B HCPCS code that is present in Part B data, CMS calculates only the payment amount under section 1847A(b)(4) of the Act per 30-day equivalent supply for that NDC-11 as set forth in paragraph § 429.425.
(C) If an NDC-11 is both present in PDE data and is associated with a Part B HCPCS code that is present in Part B data, CMS calculates both the sum of the plan-specific enrollment weighted amount per 30-day equivalent supply and the payment amount under section 1847A(b)(4) of the Act per 30-day equivalent supply for that NDC-11.
(1)
If an NDC-11 is present in both PDE data and associated with a HCPCS code present in Part B data, CMS treats each version as a distinct NDC-11 in this step of the calculation.
(ii) For each NDC-11 identified in paragraph (a)(1)(i)(A) of this section present in PDE data, CMS calculates the sum of the plan-specific enrollment weighted amounts per 30-day equivalent supply, as set forth in § 429.420(b)(1) through (8), and the total 30-day equivalent supply.
(iii) For each NDC-11 identified in paragraph (a)(1)(i)(B) of this section associated with a HCPCS present in Part B data, CMS calculates the payment amount under section 1847A(b)(4) of the Act per 30-day equivalent supply, as set forth in § 429.425(a)(4), and the total 30-day equivalent supply.
(2) CMS calculates a combined sum of the weighted averages of the sum of the plan-specific enrollment weighted amounts and the payment amounts under section 1847A(b)(4) of the Act per 30-day equivalent supply for these NDC-11s, using the respective utilization for the NDC-11 in Part B and Part D as described in the following steps:
(i) For each NDC-11 identified in paragraphs (a)(1)(ii) and (iii) of this section, CMS divides the total 30-day equivalent supply for that NDC-11 by the total 30-day equivalent supply across all NDC-11s (both those present in PDE data and those associated with HCPCS present in Part B data) of the selected drug, and multiplies this quotient by either the sum of the plan-specific enrollment weighted amounts or the payment amount under section 1847A(b)(4) of the Act, respectively, per 30-day equivalent supply of that NDC-11.
(A) If an NDC-11 is present in both PDE data and associated with a HCPCS code present in Part B data, CMS treats each version as a distinct NDC-11 in this step of the calculation.
(ii) CMS sums the amounts calculated in paragraph (a)(2)(i) of this section, across all NDC-11s of the selected drug (including both the Part B version and the Part D version of NDC-11s that are present in both PDE records associated with HCPCS codes present in Part B data), to generate the combined sum of the plan-specific enrollment weighted amounts and payment amount under section 1847A(b)(4) of the Act for the selected drug for a 30-day equivalent supply.
(3) Paragraphs (a)(2)(i) and (ii) of this section, result in the combined sum of the plan-specific enrollment weighted amounts and payment amounts under section 1847A(b)(4) of the Act across all NDC-9s of the selected drug.
(b)
Suggestion of Error.
A Primary Manufacturer that believes in good faith
( printed page 36355)
that CMS has made an error in the calculations specified in paragraph (a) of this section may submit a Suggestion of Error, as set forth in § 429.445(c) and in a form and manner specified by CMS, as set forth in § 429.445(d).
Determination of the applicable average non-FAMP amounts and applicable percent of the average non-FAMP.
(a)
Determination of the average non-FAMP.
CMS calculates and determines the lower of the two applicable average non-FAMP amounts, as set forth in paragraphs (a)(1) and (a)(2) of this section, and determines the applicable percent of the average non-FAMP to apply, as set forth in paragraph (a)(4) of this section, for all selected drugs, subject to § 429.440(b)(4), if applicable.
(1)
Average non-FAMP in calendar year 2021.
As described in section 1194(c)(1)(C)(ii)(I) of the Act, CMS calculates an amount equal to the applicable percent, with respect to the selected drug, of the average non-FAMP in calendar year 2021 (or for the first full year following market entry for such drug if there is not a non-FAMP for such drug or an average non-FAMP cannot be calculated), increased by the percentage increase in the CPI-U from September 2021 (or December of such first full year following the market entry), as applicable, to September of the year that is 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation.
(i) As set forth in § 429.100(f), CMS uses NDC-11s from the list of NDC-11s of the selected drug as set forth in paragraph § 429.100(c) to determine which NDC-11s of the selected drug are included in the ceiling calculations for the calendar year as set forth in (a)(1) of this section that meet the following criteria:
(A) The NDC-11 is assigned to the Primary Manufacturer or manufactured, marketed, controlled, or sold by Secondary Manufacturer(s);
(B) The NDC-11 does not represent a sample package;
(C) CMS received non-FAMP data for the NDC-11 for at least one calendar quarter in calendar year as set forth in paragraph (a)(1) of this section; and
(D) CMS observes any PDE days' supply and PDE quantity dispensed or any Part B data associated with the HCPCS code to which the NDC-11 is assigned in calendar year as set forth in paragraph (a)(1) of this section.
(E) The PDE record meets the inclusion and exclusion criteria as set forth in § 429.120(a)(2) through (a)(5) or the Part B data meets the inclusion and exclusion criteria as set forth in § 429.120(b)(1)(ii) through (vi) and (b)(2)(ii) through (ix).
(ii) CMS uses the non-FAMP price and unit volume data for each NDC-11 that meets the criteria as set forth in paragraph (a)(1)(i) of this section, to be included in this average non-FAMP calculation.
(iii) CMS uses the data that is submitted by the Primary Manufacturer in accordance with section 1193(a)(4)(A) of the Act, as set forth in § 429.405, for each quarter of calendar year 2021 to calculate an annual average non-FAMP per unit for calendar year set forth in paragraph (a)(1) of this section.
(iv) CMS uses 2021 PDE quantity dispensed and days' supply data (or data for the first full year following market entry for such drug if such data is not available for 2021) submitted to CMS at the NDC-11 level by Part D plan sponsors, and total Part B billing units from Part B data and 30-day equivalent supply data at the HCPCS code-level, as applicable, to calculate:
(A) An annual average non-FAMP per unit for each NDC-11 of the selected drug.
(B) The annual average non-FAMP per 30-day equivalent supply for each NDC-11 of the selected drug.
(C) The annual average non-FAMP per 30-day equivalent supply for the selected drug.
(2)
Average non-FAMP for the calendar year that is 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation.
As described in section 1194(c)(1)(C)(ii)(II) of the Act, CMS calculates an amount equal to the applicable percent, with respect to the selected drug, of the average non-FAMP for the year prior to the selected drug publication date with respect to such initial price applicability year (that is, for the calendar year that is 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation).
(i) As set forth in § 429.100(f), CMS uses NDC-11s from the list of NDC-11s of the selected drug as set forth in paragraph § 429.100(c) to determine which NDC-11s of the selected drug are included in the ceiling calculations for the calendar year as set forth in (a)(2) of this section that meet the following criteria:
(A) The NDC-11 is assigned to the Primary Manufacturer or manufactured, marketed, controlled, or sold by a Secondary Manufacturer(s);
(B) The NDC-11 does not represent a sample package;
(C) CMS received non-FAMP data for the NDC-11 for at least one calendar quarter during the calendar year and as set forth in paragraph (a)(2) of this section; and
(D) CMS observes any PDE days' supply and PDE quantity dispensed or any Part B data associated with the HCPCS code to which the NDC-11 is assigned in the calendar year as set forth in paragraph (a)(2) of this section.
(E) The PDE record meets the inclusion and exclusion criteria as set forth in § 429.120(a)(2) through (a)(5) or the Part B data meets the inclusion and exclusion criteria as set forth in § 429.120(b)(1)(ii) through (vi) and (b)(2)(ii) through (ix).
(ii) CMS uses the non-FAMP price and unit volume data for each NDC-11 that meets the criteria as set forth in paragraph (a)(2)(i) of this section, to be included for the calendar year set forth in paragraph (a)(2) of this section using the same methodology described in paragraphs (a)(1)(ii) through (iv) of this section to calculate the average non-FAMP.
(3)
Applicable percent of the average non-FAMP.
For each calendar year set forth in paragraphs (a)(1) and (a)(2) of this section, CMS calculates:
(i) The applicable percent of the average non-FAMP for each NDC-11 of the selected drug as a 30-day equivalent supply (in accordance with the methodology described in § 429.415).
(ii) The applicable percent of the average non-FAMP across the NDC-11s of the selected drug as a 30-day equivalent supply.
(4) To determine the average non-FAMP for each NDC-11 and across all NDC-11s of the selected drug, CMS conducts the following steps separately for the calendar years set forth in paragraphs (a)(1) and (a)(2) of this section:
(i) For each NDC-11 and for each quarter during the calendar year, CMS calculates the non-FAMP per unit by dividing the non-FAMP per package by the total number of NCPDP units per package.
(A) For the calendar year set forth in paragraph (a)(1) of this section only, if the non-FAMP is missing for all NDC-11s of the selected drug for such calendar year, as set forth in § 429.405, then CMS uses the non-FAMP for the quarters of the first full calendar year following the market entry for such drug.
(ii) For each NDC-11 and for each quarter during the calendar year, CMS divides the total unit volume (calculated as the product of the total number of packages sold from manufacturer-reported non-FAMP data and the number of units per package) in that quarter by the total unit volume across all four quarters during the calendar
( printed page 36356)
year (also calculated from manufacturer-reported non-FAMP data), and multiply this quotient by the non-FAMP per unit calculated in paragraph (a)(4)(i) of this section.
(A) For the calendar year set forth in paragraph (a)(1) of this section only, if the non-FAMP is missing for all NDC-11s of the selected drug for such calendar year, as set forth in § 429.405, then CMS uses the non-FAMP and total unit volumes for the quarters of the first full calendar year following the market entry for such drug.
(iii) For each NDC-11, CMS sums the amounts calculated in paragraph (a)(4)(ii) of this section across quarters to calculate the average non-FAMP per unit for that NDC-11 for the calendar year.
(iv) For each NDC-11, CMS divides the total units for that NDC-11 by the total units for all applicable NDC-11s and multiplies this quotient by the average non-FAMP per unit for the calendar year calculated in paragraph (a)(4)(iii) of this section, applying the methodology that follows, as applicable:
(A) For NDC-11s that are present only in PDE data: Total units are defined as the total quantity dispensed for an NDC-11 as determined using the applicable calendar year as set forth in paragraphs (a)(1) and (a)(2) of this section and PDE data identified in § 429.410(b)(5)(iii) and (iv).
(B) For NDC-11s that are only associated with HCPCS codes present in Part B data: Total units are defined as the total Part B units for an NDC-11 as determined using the unit allocation and standardization methodology described for calculating converted Part B units attributed to the NDC-11 as set forth in § 429.425(a)(3).
(C) For NDC-11s that are both present in PDE data and associated with HCPCS codes present in Part B data: Total units are defined as the sum of the total quantity dispensed for the NDC-11 and the total Part B units for an NDC-11 as determined using the unit allocation and standardization methodology described in § 429.425(a)(3).
(v) For the calendar year as set forth in paragraph (a)(1) of this section only: for each NDC-11, CMS then increases the average non-FAMP per unit calculated in paragraph (a)(4)(iv) of this section by the percentage increase in CPI-U (all items; United States city average) as set forth in paragraph (a)(1) of this section.
(A) Notwithstanding paragraph (a)(4)(v) of this section, CMS does not apply a CPI-U adjustment to the average non-FAMP per unit for the calendar year described in paragraph (a)(2) of this section.
(vi) For each NDC-11, after CMS calculates the average non-FAMP per unit for the calendar year (as calculated in paragraph (a)(4)(iv) of this section for the calendar year as set forth in paragraph (a)(2) of this section and as calculated in paragraph (a)(4)(v) of this section for the calendar year as set forth in paragraph (a)(1) of this section), CMS applies the applicable percent specified in section 1194(c)(3) of the Act for the monopoly type which are set forth in paragraphs (A)(1) through (3) of this section and are determined for the selected drug based on its initial approval date as set forth in § 429.125(a)(1)(i) and the initial price applicability year for which the drug is selected for negotiation.
(A) The monopoly types and applicable percent are:
(1)
A short-monopoly drug or vaccine, as described in section 1194(c)(3)(A) of the Act, is a selected drug other than an extended-monopoly drug and a long-monopoly drug, for which the applicable percent is 75 percent.
(2)
An extended-monopoly drug, as described in section 1194(c)(3)(B) of the Act and defined in § 429.20, for which the applicable percent is 65 percent.
(3)
A long-monopoly drug, as described in section 1194(c)(3)(C) of the Act and defined in § 429.20, for which the applicable percent is 40 percent.
(vii) For each NDC-11, CMS then multiplies the average non-FAMP per unit for the calendar year, adjusted for inflation, if applicable per paragraph (a)(4)(v) of this section, and with the applicable percent applied as determined in paragraph (a)(4)(vi) of this section by the quotient of total units divided by the total 30-day equivalent supply.
(viii) CMS sums the amounts calculated in paragraph (a)(4)(vii) of this section, across all NDC-11s of the selected drug to calculate the average non-FAMP per 30-day equivalent supply for the calendar year, adjusted for inflation, if applicable per paragraph (a)(4)(v) of this section, and with the applicable percent applied as specified in paragraph (a)(4)(vi) of this section, for the selected drug.
(ix) CMS compares the amount calculated in paragraph (a)(4)(viii) of this section for the applicable percent of the calendar year described in paragraph (a)(1) of this section average non-FAMP per 30-day equivalent supply for the calendar year, adjusted for inflation, with the amount calculated in paragraph (a)(4)(viii) of this section for the applicable percent of the calendar year described in paragraph (a)(2) of this section average non-FAMP per 30-day equivalent supply for the calendar year and determines which is lower.
(b)
Suggestion of Error.
A Primary Manufacturer that believes in good faith that CMS has made an error in the calculations specified in paragraph (a) of this section may submit a Suggestion of Error, as set forth in § 429.445(c) and in a form and manner specified by CMS, as set forth in § 429.445(d).
(a)
Definitions.
For the purposes of this section, the following definitions apply:
(1)
Part B 2021 Manufacturer
means the NDA holder or the BLA holder for the qualifying single source drug on December 31, 2021.
(2)
Part D 2021 Manufacturer
means the entity that either (1) had a Medicare Coverage Gap Discount Program (CGDP) Agreement under section 1860D-14A of the Act in effect for the qualifying single source drug on December 31, 2021, or (2) had an arrangement whereby the manufacturer's labeler codes were listed on another manufacturer's Medicare CGDP Agreement, consistent with section 1860D-14A of the Act, in effect on December 31, 2021.
(3)
Part B 2021 Manufacturer and its controlled group
comprises all persons that, as of December 31, 2021, were treated as a single employer under subsection (a) or (b) of section 52 of the Internal Revenue Code of 1986 with the Part B 2021 Manufacturer.
(4)
Part D 2021 Manufacturer and its controlled group
comprises all persons that, as of December 31, 2021, were treated as a single employer under subsection (a) or (b) of section 52 of the Internal Revenue Code of 1986 with the Part D 2021 Manufacturer and had a CGDP Agreement in effect on December 31, 2021.
(b)
General rule.
For a drug selected for negotiation for initial price applicability year 2029 or 2030, or a drug selected for renegotiation for initial price applicability year 2029 or 2030, for which the Primary Manufacturer submits information in accordance with paragraph (b)(1) of this section and CMS determines that such drug meets the requirements set forth in paragraph (b)(2) of this section, CMS will not make an offer (or agree to a counteroffer) for an MFP (as described in § 429.500(b)) that is lower than the Temporary Floor for Small Biotech Drugs, which must be equal to the amount specified in paragraph (b)(3) of this section.
(1)
Temporary Floor for Small Biotech Drugs submission.
For a selected drug to be considered for eligibility for the Temporary Floor for Small Biotech
( printed page 36357)
Drugs, such drug's Primary Manufacturer must submit information to CMS regarding the eligibility criteria described in paragraph (b)(2) of this section as relevant to such drug, in the form and manner specified by CMS.
(2)
Eligibility for the Temporary Floor for Small Biotech Drugs.
To be eligible for the Temporary Floor for Small Biotech Drugs, CMS must determine either that the selected drug is covered under Part D and meets the requirements of the Part D Track set forth in paragraph (b)(2)(i) of this section or that the selected drug is payable under Part B and meets the requirements of the Part B Track set forth in paragraph (b)(2)(ii) of this section.
(i)
Part D Track.
To meet the requirements of the Part D Track, the total expenditures under Part D during 2021 for the selected drug (calculated using the methodology set forth in § 429.120(a), except the date of service as described in § 429.120(a)(1) is during 2021) must be:
(A) Equal to or less than 1 percent of the total expenditures under Part D for all covered Part D drugs during 2021; and
(B) Equal to or greater than 80 percent of the total expenditures under Part D for all covered Part D drugs during 2021 for which the Part D 2021 Manufacturer and its controlled group had an agreement in effect under section 1860D-14A on December 31, 2021.
(ii)
Part B Track.
To meet the requirements of the Part B Track, the total expenditures under Part B during 2021 for the selected drug calculated using the methodology set forth in § 429.120(b), except the date of service as described in § 429.120(b)(1)(i) and (b)(2)(i) is during 2021, must be:
(A) Equal to or less than 1 percent of the total expenditures under Part B for all qualifying single source drugs payable under Part B during 2021; and
(B) Equal to or greater than 80 percent of the total expenditures under Part B during 2021 for all qualifying single source drugs of the Part B 2021 Manufacturer and its controlled group that are payable under Part B during 2021.
(iii)
Limitation.
A selected drug will not be eligible for the Temporary Floor for Small Biotech Drugs if the Primary Manufacturer of such drug was acquired after 2021 by another manufacturer that does not meet the definition of a specified manufacturer under section 1860D-14C(g)(4)(B)(ii) of the Act, effective at the beginning of the plan year immediately following such acquisition or, in the case of an acquisition before 2025, effective January 1, 2025.
(3)
Determination of the Temporary Floor for Small Biotech Drugs.
CMS calculates the Temporary Floor for Small Biotech Drugs with respect to each selected drug that CMS determines to be eligible, under paragraph (b)(2) of this section, as 66 percent of the following amount, as applicable:
(i) Subject to paragraph (b)(3)(ii) of this section, the average non-FAMP in calendar year 2021, calculated using the methodology set forth in § 429.435, increased by the percentage increase in the CPI-U from September 2021 to September of the year prior to the selected drug publication date for which the drug is selected for negotiation or, as applicable, renegotiation.
(ii) In the case that there is not an average non-FAMP available for such drug for 2021, the average non-FAMP for the first full year following market entry, increased by the percentage increase in the CPI-U from December of such first full year following market entry to September of the year prior to the selected drug publication date for which the drug is selected for negotiation or, as applicable, renegotiation.
(4)
Determination of the adjusted ceiling for the MFP exception.
In the event that the ceiling identified in § 429.410(b) (or § 429.620(b) as applicable) for a Small Biotech Drug is below the Temporary Floor for Small Biotech Drugs for such Small Biotech Drug, as described in paragraph (b)(3) of this section, then, notwithstanding § 429.410 (or § 429.620 as applicable), CMS will not make an offer (or agree to a counteroffer) for an MFP that exceeds the adjusted ceiling determined under paragraphs (b)(4)(i) or (ii) of this section, as applicable, and calculates such adjusted ceiling amount as follows:
(i) CMS excludes the non-FAMP ceiling amount identified under sections 1194(c)(1)(C)(i) or 1194(c)(1)(C)(ii)(I) of the Act from the ceiling determination made under § 429.410(b) or § 429.620(b), as applicable, with respect to such selected drug. If the resulting ceiling amount is equal to or above the Temporary Floor for Small Biotech Drugs established in paragraph (b)(3) of this section, then, notwithstanding § 429.410 or § 429.620(b), such resulting ceiling amount applies as the ceiling.
(ii) If the adjusted ceiling determined under paragraph (b)(4)(i) of this section is below the Temporary Floor for Small Biotech Drugs established in paragraph (b)(3) of this section, then, notwithstanding § 429.410 or § 429.620(b), the adjusted ceiling equals the Temporary Floor for Small Biotech Drugs established in paragraph (b)(3) of this section.
(5)
Notice of determination.
After review of an application made in accordance with paragraph (b)(1) of this section, CMS provides a notice in writing to the Primary Manufacturer, alongside the calculation information provided as set forth in § 429.445(a), of:
(i) A determination of whether the selected drug is a Small Biotech Drug; and
(ii) If the selected drug is eligible for the Temporary Floor for Small Biotech Drugs, the calculation of the Temporary Floor for Small Biotech Drugs set forth in § 429.440(b)(3) and the calculation of the ceiling (or adjusted ceiling, if applicable, set forth in § 429.440(b)(4).
(6)
Suggestion of Error.
A Primary Manufacturer that believes in good faith that CMS has made an error in the calculations specified in paragraph (b)(3) or (b)(4) of this section may submit a Suggestion of Error, as set forth in § 429.445(c) and in a form and manner specified by CMS, as set forth in § 429.445(d).
(a)
Calculation Information.
CMS provides a Primary Manufacturer with the following, as applicable:
(1) Information on CMS' calculation of the ceiling, as described in §§ 429.410 through 429.435;
(2) The computation of how CMS will apply a single MFP across dosage forms and strengths of the selected drug, as described in § 429.700(b) and (c); and
(3) Information on CMS' calculation of the Temporary Floor for Small Biotech Drugs (and adjusted ceiling, as applicable), as described in § 429.440(b)(3) and (b)(4).
(b)
Timing.
(1) CMS intends to provide the information specified in paragraphs (a)(1), (a)(2), and (a)(3) (as applicable) of this section:
(i) Following the Primary Manufacturer's submission of data set forth in §§ 429.100(d), 429.405(a), 429.440(b)(1), and 429.505(b)(2); and
(ii) Following the Primary Manufacturer's submission of data set forth in § 429.615(b)(1).
(2) CMS intends to provide the information specified in paragraph (a)(2) of this section:
(i) Following the Primary Manufacturer's submission of data set forth in § 429.100(e); and
(ii) Following the determination by CMS that an NDC with insufficient data, as described in proposed § 429.700(c)(3), has sufficient data as described in § 429.700(c)(4)(i)(B)(2).
( printed page 36358)
(c)
Suggestion of Error.
A Primary Manufacturer that believes in good faith that CMS has made an error in the calculations specified in paragraphs (a)(1), (a)(2), or (a)(3) of this section may submit a Suggestion of Error pertaining to such calculations for CMS' consideration.
(1) A Primary Manufacturer must submit a Suggestion of Error within 10 days of receiving information under paragraph (a) of this section from CMS.
(2) The Suggestion of Error process does not affect a Primary Manufacturer's obligation to comply with Negotiation Program requirements and does not alter or change any timelines or requirements of the Negotiation Program.
(d)
Method and process.
A Primary Manufacturer must submit any Suggestion of Error, as described in paragraph (c) of this section, in a form and manner specified by CMS.
(a) CMS has a consistent methodology and process for negotiating MFPs as set forth in this subpart. For purposes of negotiating or renegotiating the MFP of a selected drug in accordance with such methodology and process, CMS:
(1) Aims to achieve agreement on the lowest MFP for each selected drug consistent with section 1194(b)(1) of the Act; and
(2) Considers the factors set forth in section 1194(e) of the Act and § 429.505, as applicable to the selected drug, in determining offers and counteroffers.
(b) To formalize agreement on an MFP, CMS and the Primary Manufacturer both must sign an Addendum to the Negotiation Program Agreement as described in § 429.200(e) that sets forth the agreed-upon MFP.
(a) For purposes of negotiating the maximum fair price of a selected drug with the Primary Manufacturer, CMS considers the Primary Manufacturer-required data specified in § 429.505(b)(2) and evidence about the selected drug and therapeutic alternatives specified in § 429.505(d)(3), as applicable to the drug, as the basis for determining the offers and counteroffers for the selected drug.
(b)
Primary Manufacturer-required data.
(1) In accordance with the requirements of the Negotiation Program Agreement as set forth in § 429.200, the Primary Manufacturer must submit the information in paragraph (b)(2) of this section to CMS in a form and manner specified by CMS by 11:59 p.m. PST on March 1 of the year of the selected drug publication date for the selected drug, inclusive of the NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer.
(2) Primary Manufacturer required data includes—
(i) Research and development costs of the Primary Manufacturer for the selected drug and the extent to which the Primary Manufacturer has recouped research and development costs;
(ii) Current unit costs of production and distribution of the selected drug;
(iii) Prior Federal financial support for novel therapeutic discovery and development with respect to the selected drug;
(iv) Data on pending and approved patent applications, exclusivities recognized by the Food and Drug Administration (FDA), and applications and approvals under section 505(c) of the FD&C Act (21 U.S.C. 355(c) or section 351(a) of the PHS Act (42 U.S.C. 262(a)) for the selected drug; and
(v) Market data and revenue and sales volume data for the selected drug in the United States.
(c) In accordance with the requirements of the Negotiation Program Agreement as set forth in § 429.200, a Primary Manufacturer is required to provide updates to the information described in paragraph (b)(2) of this section for the drug selected for negotiation in a form and manner prescribed by CMS if the Primary Manufacturer becomes aware that any of such information has changed or is otherwise inaccurate.
(d)
Evidence about the selected drug and therapeutic alternatives—
(1) Any interested party, including the Primary Manufacturer of a selected drug, may choose to submit the information set forth in paragraph (d)(3) of this section to CMS in a form and manner specified by CMS.
(2) Interested parties must submit the information in paragraph (d)(3) of this section by 11:59 p.m. PST on March 1 of the year of the selected drug publication date for a selected drug in a form and manner specified by CMS.
(3) Evidence about the selected drug and therapeutic alternatives includes—
(i) The extent to which a selected drug represents a therapeutic advance as compared to existing therapeutic alternatives and the costs of such existing therapeutic alternatives;
(ii) Prescribing information approved by the FDA for a selected drug and therapeutic alternatives to such selected drug;
(iii) Comparative effectiveness of a selected drug and therapeutic alternatives to such selected drug, taking into consideration the effects of the selected drug and therapeutic alternatives to such selected drug on specific populations, such as individuals with disabilities, the elderly, the terminally ill, children, and other patient populations; and
(iv) The extent to which a selected drug and therapeutic alternatives to such selected drug address unmet medical needs for a condition for which treatment or diagnosis is not addressed adequately by available therapy.
(e)
Limitation on evidence.
CMS will not use evidence from comparative clinical effectiveness research in a manner that treats extending the life of an elderly, disabled, or terminally ill individual as of lower value than extending the life of an individual who is younger, nondisabled, or not terminally ill.
(1) CMS reviews cost-effectiveness measures used in studies relevant to a selected drug to determine whether the use of the measure is permitted in accordance with section 1194(e)(2) of the Act as described at paragraph (e) of this section, as well as section 1182(e) of the Act and other applicable law.
(2) CMS may use content in a study that uses a cost-effectiveness measure if it determines that the cost-effectiveness measure used is permitted in accordance with section 1194(e)(2) of the Act as described at paragraph (e) of this section, as well as section 1182(e) of the Act and other applicable law.
(a)
Identification of conditions for which the selected drug is used.
CMS identifies the condition(s) for which the selected drug is used that are covered under Part D, payable under Part B, or both.
(1) CMS considers the prescribing information for the selected drug in accordance with section 1194(e)(2)(B) of the Act.
(2) CMS may consider off-label use as defined in § 429.20.
(3) CMS excludes FDA-approved indications and off-label use, if applicable, from its analysis when CMS believes that utilization of the selected drug within such indication(s) and for such uses is intended solely for use in a setting in which the selected drug is not payable under Part B and not covered under Part D.
(b)
Information used to identify a therapeutic alternative(s) for the selected drug.
(1) CMS identifies the therapeutic alternative(s) for each condition(s) for which the selected drug is used as
( printed page 36359)
determined under § 429.510(a) using any combination of the following, as available:
(i) Information submitted by the Primary Manufacturer and the public as described in § 429.505(b)(2) and (d)(3);
(ii) Prescribing information as approved by the FDA;
(iii) Drug classification systems commonly used in the public and private sector for formulary development;
(iv) Major drug compendia;
(v) Widely accepted clinical practice guidelines;
(vi) Evidence identified through a CMS-led literature review;
(vii) Published drug or drug class reviews;
(viii) Peer-reviewed studies or other clinical evidence and information submitted by the Primary Manufacturer and the public; or
(ix) Medicare claims or other data sets.
(2) The therapeutic alternative(s) may be a brand name drug, a brand name biological product, a generic drug, or a biosimilar, including specific formulations or dosage forms and strengths of such brand name drug, brand name biological product, generic drug, or biosimilar.
(c)
Process for determining the potential therapeutic alternative(s) for a selected drug.
(1) CMS may consult with FDA, clinicians, patients or patient organizations, and researchers.
(2) When determining a therapeutic alternative(s) for a selected drug, CMS considers:
(i) Off-label use(s) for potential therapeutic alternatives as defined in § 429.20;
(ii) Potential therapeutic alternatives within the same pharmacologic class as the selected drug based on properties such as chemical class, therapeutic class, or mechanism of action;
(iii) Potential therapeutic alternatives in a different pharmacologic class than the selected drug; and
(iv) Formulations or dosage forms and strengths of a potential therapeutic alternative.
(3) CMS may focus on a subset of therapeutic alternatives that are clinically comparable to the selected drug.
(4) CMS prioritizes clinical appropriateness when identifying the therapeutic alternative(s) for a selected drug.
(d)
Determining a starting point for the initial offer.
(1) CMS determines the price of each therapeutic alternative determined under paragraph (c) of this section:
(i) For a therapeutic alternative covered under Part D, the price is the lower of:
(A) The Net Part D Plan Payment and Beneficiary Liability of the therapeutic alternative;
(B) The MFP of the therapeutic alternative, if applicable; or
(C) The WAC of the therapeutic alternative.
(ii) For a therapeutic alternative payable under Part B, the price is the lower of:
(A) The ASP of the therapeutic alternative;
(B) The MFP of the therapeutic alternative, if applicable; or
(C) The WAC of the therapeutic alternative.
(iii) For a therapeutic alternative that is both covered under Part D and payable under Part B, CMS combines the prices determined in paragraphs (d)(1)(i) and (d)(1)(ii) of this section using an approach similar to the methodology used to combine the sum of the plan-specific enrollment weighted amounts and the payment amount under section 1847A(b)(4) of the Act to calculate the combined Part B and Part D amount, as described in § 429.430.
(2) The prices determined in accordance with paragraph (d)(1) of this section will be expressed as a 30-day equivalent supply.
(i) For a therapeutic alternative(s) covered under Part D, the 30-day equivalent supply will be calculated as described in § 429.415(a)(1), unless CMS determines it is appropriate to apply an alternative methodology under paragraph (d)(2)(iv) of this section.
(ii) For a therapeutic alternative(s) payable under Part B, the 30-day equivalent supply will be calculated as described in § 429.415(a)(2), unless CMS determines it is appropriate to apply an alternative methodology under paragraph (d)(2)(iv) of this section.
(iii) For a therapeutic alternative(s) payable under Part B and covered under Part D, the 30-day equivalent supply will be calculated separately for the prices determined in paragraphs (d)(1)(i) and (d)(1)(ii) of this section using the methodology described in paragraphs (d)(2)(i) and (d)(2)(ii) of this section, unless CMS determines it is appropriate to apply an alternative methodology under paragraph (d)(2)(iv) of this section. The separate prices expressed as a 30-day equivalent supply for the therapeutic alternative as payable under Part B and as covered under Part D are then combined using the methodology described in paragraph (d)(1)(iii) of this section to result in one combined price expressed as a 30-day equivalent supply for the therapeutic alternative.
(iv) For a therapeutic alternative(s) covered under Part D, payable under Part B, or both, for which CMS has determined it is appropriate to apply an alternative methodology, the 30-day equivalent supply may be calculated using a tailored methodology that promotes comparability between the therapeutic alternative(s) and the selected drug.
(3) CMS uses the price(s) determined in paragraph (d)(1) of this section to determine the starting point for developing the initial offer.
(i) If there is no therapeutic alternative for the selected drug or if there is no price(s) of the therapeutic alternative(s) determined under paragraph (d)(1) of this section below the ceiling determined under § 429.410(b), then the starting point for developing the initial offer is the lower of:
(A) The pharmaceutical price for the selected drug as included in the Federal Supply Schedule as managed by the Department of Veterans Affairs per 48 CFR part 38 as most recently submitted by the Primary Manufacturer under § 429.505(b);
(B) The maximum price a manufacturer can charge for a selected drug under 38 U.S.C. 8126 as most recently submitted by the Primary Manufacturer under § 429.505(b); or
(C) The ceiling for the selected drug as determined in § 429.410(b).
(ii) If there is more than one therapeutic alternative for the selected drug, and at least one price determined under paragraph (d)(1) of this section is below the ceiling determined under § 429.410(b), CMS determines a starting point for developing the initial offer within a range based on the lower of the prices listed in paragraph (d)(1) of this section and the ceiling determined under § 429.410(b).
(iii) If there is one therapeutic alternative for the selected drug with a price that is below the ceiling, the price of such therapeutic alternative is the starting point.
(e)
Adjusting the starting point based on section 1194(e)(2) factors.
(1) CMS uses a qualitative approach to broadly evaluate the body of available evidence related to section 1194(e)(2) factors listed at § 429.505(d)(3) in totality, including any combination of the following, as available:
(i) Information submitted by the Primary Manufacturer and the public as described in § 429.505(b)(2) and (d)(3) and provided in public events described in § 429.515(b), as available;
(ii) Evidence identified through a CMS-led literature review;
( printed page 36360)
(iii) Medicare claims or other datasets, potentially including evidence related to health care resource utilization and usage patterns of the selected drug and its therapeutic alternative(s) as determined in paragraph (c) of this section, if any;
(iv) Clinical data;
(v) Other information relevant to the selected drug and its therapeutic alternative(s), if any.
(2) CMS may consult with FDA, clinicians, patients or patient organizations, and researchers.
(3) CMS identifies outcomes of interest to evaluate for each condition identified under § 429.510(a).
(i) The types of outcomes and contextual factors that CMS considers include:
(A) Clinical outcomes;
(B) Patient-centered outcomes, patient experience data, and patient-reported outcomes, if available;
(C) Additional outcomes and contextual factors or patient and caregiver preferences to the extent these outcomes and factors correspond with benefits or harms to individuals taking the selected drug or therapeutic alternative(s), if any; and
(D) Caregiver perspective to the extent that such perspective reflects directly upon the experience or relevant outcomes of the patient taking the selected drug.
(ii) CMS identifies outcomes using:
(A) A CMS-led literature review; and
(B) Information submitted by the Primary Manufacturer and the public as described in § 429.505(b)(2) and (d)(3) and provided in public events described in § 429.515(b), as available.
(4) Based on the evaluation of evidence described in paragraphs (e)(1) through (3) of this section, CMS adjusts the starting point by considering the applicable evidence related to section 1194(e)(2) factors collectively and within the context of the course of care for each condition for which the selected drug is used.
(i) This assessment includes but is not limited to examining the improvements in outcomes to determine the extent to which a selected drug represents a therapeutic advance as compared to its therapeutic alternative(s) by considering:
(A) The costs of the selected drug and its therapeutic alternative(s); and
(B) The magnitude of differences in outcomes of interest determined in paragraph (e)(3) of this section conferred by the selected drug compared to its therapeutic alternative(s) for each condition for which CMS has identified a therapeutic alternative.
(ii) Notwithstanding paragraph (e)(4)(i) of this section, if a condition identified under paragraph (a) of this section does not have a therapeutic alternative as identified under paragraphs (b) and (c) of this section, then CMS considers:
(A) The totality of available information described in paragraph (e)(1) of this section;
(B) Any existing treatment option(s) to determine the extent to which the selected drug addresses an unmet medical need as defined in § 429.20 for each identified condition; and
(C) The magnitude of differences in outcomes of interest determined in paragraph (e)(3) of this section conferred by the selected drug to determine the extent to which the selected drug represents a therapeutic advance.
(5) The starting point may be adjusted upward, adjusted downward, or not adjusted based on considerations described in paragraphs (e)(1) through (4) of this section to determine the preliminary price.
(f)
Adjusting the preliminary price based on section 1194(e)(1) factors.
(1) CMS considers the section 1194(e)(1) factors, specified in § 429.505(b)(2) and provided by the Primary Manufacturer, in totality in determining whether to adjust the preliminary price.
(2) The preliminary price may be adjusted upward, adjusted downward, or not adjusted based on CMS' consideration of the section 1194(e)(1) factors in totality as described in paragraph (f)(1) of this section.
(3) The price resulting from paragraphs (f)(1) and (f)(2) of this section is the initial offer.
(i) Notwithstanding paragraph (f)(3) of this section, if the amount determined in accordance with the process described in this section is above the ceiling as determined in § 429.410(b), then the initial offer will be equal to the ceiling, and if the amount determined in accordance with this section is below the temporary floor for small biotech drugs (as determined in § 429.440(b)), if applicable, then the initial offer will be equal to the temporary floor.
Engagement with Primary Manufacturers and interested parties.
(a)
Engagement with Primary Manufacturers.
In a form and manner specified by CMS and in accordance with the rules set forth in this § 429.515, CMS hosts up to four optional meetings with Primary Manufacturers of selected drugs that have submitted the information set forth in § 429.505.
(1)
Meeting scope.
The first meeting that CMS offers Primary Manufacturers the option to attend is intended for the Primary Manufacturer to provide additional context on their data submission of the section 1194(e)(1) factors and section 1194(e)(2) factors as set forth in § 429.505 as CMS begins evaluating the data submission and developing an initial offer as described in § 429.510.
(i) The remaining three meetings that CMS offers Primary Manufacturers the opportunity to attend focus on the section 1194(e)(1) factors and section 1194(e)(2) factors described in § 429.505 and other topics aimed at working towards an agreement on an MFP.
(ii) Notwithstanding paragraph (a)(1) of this section, new manufacturer-required data as described in § 429.505(b)(2) is not considered if it is submitted after the March 1 deadline.
(iii) Discussion of disputes and program policies regarding the negotiation process are considered out of scope.
(2)
Meeting attendance.
Meetings are attended solely by representatives of the Primary Manufacturer and of CMS. The number of attendees are limited as specified by CMS.
(3)
Meeting materials.
For the purposes of each meeting:
(i) Primary Manufacturers may provide the following to be presented and discussed at each meeting in a form and manner specified by CMS:
(A) New information on the section 1194(e)(2) factors
(B) Materials, including pages, slides, and charts and graphs, to facilitate discussion, which must comply with limits on the amount or format of such materials as specified by CMS.
(ii) CMS may request that the Primary Manufacturer provide copies of any presented or discussed materials after the meeting in which they are presented or discussed.
(4)
Meeting timing.
The first optional meeting, if accepted by the Primary Manufacturer, occurs, at a time to be specified by CMS, after the data submission deadline specified in § 429.505(b)(1) and (d)(2) and before the provision of CMS' initial offer, as set forth in § 429.520. Up to three optional meetings are offered by CMS to occur after the provision of CMS' initial offer. If the Primary Manufacturer accepts the meeting invitation(s), such meeting(s) occur, at a time to be specified by CMS, after the provision of CMS's initial offer as set forth in § 429.520 and before the provision of CMS' final offer, if applicable, as set forth in § 429.535.
(5)
Meeting rules.
A Primary Manufacturer—
(i) May maintain a written record of the negotiation process; and
( printed page 36361)
(ii) Is prohibited from making an audio or video recording of any negotiation meeting.
(b)
Engagement with interested parties.
CMS may hold event(s) in a form and manner and at times to be specified by CMS to seek information from patients and other interested parties about selected drugs, therapeutic alternatives to the selected drugs, and other information. CMS may incorporate drugs selected for renegotiation into these events or may hold separate events, as applicable.
Provision of CMS' written initial offer and concise justification.
(a)
Written initial offer.
CMS provides the Primary Manufacturer with the written initial offer no later than June 1 following the selected drug publication date as defined in § 429.20.
(1) This written initial offer is accompanied by an Addendum to the Negotiation Program Agreement, as described in § 429.200(e), populated with the proposal for the MFP. To formalize agreement on an MFP, CMS and the Primary Manufacturer both must sign an Addendum to the Negotiation Program Agreement as described in § 429.200(e) that sets forth the agreed-upon MFP as described in § 429.500(b).
(b)
Concise justification.
With the written initial offer, CMS includes a concise justification for the written initial offer based on the data set forth in § 429.505. The concise justification:
(1) Includes a qualitative description of the factors from section 1194(e) of the Act as set forth in § 429.505 and a description of the methodology that CMS used to develop the written initial offer as set forth under subpart F of this part.
(2) Provides the Primary Manufacturer with information on the range of evidence and other information considered in accordance with section 1194(e) of the Act that CMS found compelling during the development of the written initial offer.
(3) May include information obtained through events with interested parties as described at § 429.515(b).
(a)
Primary Manufacturers' statutory written counteroffer.
The Primary Manufacturer must respond in writing within 30 days of receipt of the written initial offer from CMS by either accepting the written initial offer for the selected drug or making a statutory written counteroffer and providing a justification for such counteroffer based on the data set forth in § 429.505.
(b)
Required components.
(1) Any statutory written counteroffer must:
(i) Provide a proposal for the MFP for the selected drug and a justification for such proposal;
(ii) Respond to the justification provided in CMS' written initial offer; and
(iii) Indicate the reasons the Primary Manufacturer believes that the information submitted by the Primary Manufacturer described in § 429.505, or other available data as described in § 429.505(d), supports the Primary Manufacturer's statutory written counteroffer or otherwise does not support CMS' written initial offer.
(2) Primary Manufacturers must complete an Addendum to the Negotiation Program Agreement, as described in § 429.200(e), and must submit the statutory written counteroffer in a form and manner specified by CMS.
(c)
CMS response to statutory written counteroffer.
CMS responds in writing to a statutory written counteroffer made by the Primary Manufacturer.
(1) CMS considers the statutory written counteroffer and either accepts or rejects it in writing within 30 days of receipt of the statutory written counteroffer or within 60 days of sharing the initial offer, whichever is later.
(2) To formalize agreement on an MFP, CMS and the Primary Manufacturer both must sign an Addendum to the Negotiation Program Agreement as described in § 429.200(e) that sets forth the agreed-upon MFP as described in § 429.500(b).
(a)
Additional price exchange opportunities.
CMS and Primary Manufacturers may initiate additional, written offers and counteroffers in a form and manner specified by CMS during the period of time between CMS' rejection of the Primary Manufacturer's statutory written counteroffer, if applicable, and the parties reaching an agreement on the MFP, or at least 8 business days before the deadline by which CMS must issue the final offer, whichever is earlier, via the additional price exchange opportunities described in this section.
(1) The additional price exchange opportunities allow for the optional upload of materials which must comply with limits on the amount or format of such materials as specified by CMS, and include an optional text field to enable the offering or counteroffering party to include additional contextual information for the offer or counteroffer.
(2) Only one written offer or counteroffer per selected drug may be active at a time.
(3) An offering or counteroffering party may archive its written offer or counteroffer in the period before the other party accepts or rejects it, but not afterwards.
(4) Parties do not need to alternate making written offers and counteroffers.
(5) To formalize agreement on an MFP, CMS and the Primary Manufacturer both must sign an Addendum to the Negotiation Program Agreement as described in § 429.200(e) that sets forth the agreed-upon MFP as described in § 429.500(b).
Notification of final offer and conclusion of negotiations.
(a)
Notification of final offer.
In the event neither CMS' initial offer nor the Primary Manufacturer's statutory written counteroffer were accepted, and an MFP was not agreed to during the negotiation meetings or via the additional price exchange functionality, CMS sends the Primary Manufacturer a “Notification of Final Maximum Fair Price Offer” and an Addendum with the final offer MFP by September 30 following the selected drug publication date defined in § 429.20.
(1) If a final offer is sent, the Primary Manufacturer must respond in writing to this final offer by either accepting or rejecting the final offer by October 31 following the selected drug publication date defined in § 429.20.
(2) To formalize agreement on an MFP, CMS and the Primary Manufacturer both must sign an Addendum to the Negotiation Program Agreement as described in § 429.200(e) that sets forth the agreed-upon MFP as described in § 429.500(b).
(b)
Conclusion of negotiations.
All negotiations between CMS and the Primary Manufacturer of the selected drug must end prior to November 1 following the selected drug publication date, with respect to the initial price applicability year.
(a) With respect to initial price applicability years beginning with initial price applicability year 2029, the process of renegotiation will include the following actions.
(1) CMS identifies renegotiation-eligible drugs, if any, as described in § 429.605.
(2) CMS selects drugs for renegotiation from among such renegotiation-eligible drugs, if any, as described in § 429.610.
( printed page 36362)
(3) CMS renegotiates the MFP applicable to any such drugs selected for renegotiation in accordance with the process set forth in § 429.620.
(b)
Applicability of a renegotiated MFP.
(1) Once the Primary Manufacturer and CMS have agreed upon a renegotiated MFP under the process set forth under § 429.620 with respect to a selected drug, the Primary Manufacturer is responsible for making the renegotiated MFP for the selected drug available, in accordance with subpart I of this part, with respect to dispenses, administrations, and furnishings of the selected drug on or after January 1 of the initial price applicability year for which the selected drug is selected for renegotiation (subject to proposed § 429.135) until termination of the Negotiation Program Agreement in accordance with proposed § 429.205.
(2) CMS applies a renegotiated MFP to all formulations across dosage forms and strengths of the selected drug by applying the methodology set forth at proposed § 429.700, notwithstanding proposed § 429.700(b)(1) and (2)(i) CMS uses the initial price applicability year of the renegotiated MFP to determine the applicable year of data.
(3) The Primary Manufacturer of a selected drug with a renegotiated MFP must provide access to the initial agreed-upon MFP in accordance with section 1193(a)(3), including as described in subpart I of this part, prior to the effective date of the renegotiated MFP as specified in paragraph (b)(1) of this section.
(c)
Publication of the list of drugs selected for renegotiation.
CMS publishes the drugs selected for renegotiation with respect to an initial price applicability year, if any, consistent with § 429.100(b)(2).
(a)
For a selected drug with an agreed-upon MFP from a prior initial price applicability year
—
(1) For any selected drugs with an agreed-upon MFP from a prior initial price applicability year for which CMS does not make a determination described in § 429.135(c), CMS reviews such drugs for renegotiation eligibility in accordance with section 1194(f)(2) of the Act as described in paragraphs (b) through (d) of this section.
(b)
Selected drugs for which there is a change in monopoly status.
With respect to initial price applicability years beginning in 2029, CMS determines a selected drug to be renegotiation-eligible based on a change in monopoly status as described in paragraph (b)(1) or (2) of this section.
(1) A selected drug with an agreed-upon MFP will be determined to be a renegotiation-eligible drug if there has been a change in its monopoly status from a drug that is not an extended-monopoly drug, as defined in § 429.20, or a long-monopoly drug, as defined in § 429.20, to an extended-monopoly drug by January 1 of the initial price applicability year for which such selected drug would be selected for renegotiation.
(2) A selected drug with an agreed-upon MFP will be determined to be a renegotiation-eligible drug if there has been a change in status from a drug that is not a long-monopoly drug, as defined in § 429.20, to a long-monopoly drug by January 1 of the initial price applicability year for which such selected drug would be selected for renegotiation.
(c)
Selected drugs for which a new indication is added.
A selected drug with an agreed-upon MFP will be determined to be a renegotiation-eligible drug if a new indication, which may be a new indication added to the FDA-approved labeling or a new off label use, is added for the selected drug since the selected drug was last negotiated or renegotiated, as applicable.
(1) A selected drug will not be determined to be renegotiation-eligible based on the addition of a new indication if the FDA-approved labeling update or off-label use is related to a previously indicated condition.
(2) CMS determines if a new indication has been added as described in paragraph (c)(1) of this section by evaluating:
(i) FDA-approved labeling for the selected drug; and
(ii) Voluntary submissions from the Primary Manufacturer of the selected drug as described in § 429.615, if any.
(A) If a Primary Manufacturer voluntarily submits information through the process described in § 429.615, then CMS may consider off-label use when determining if a new indication has been added for renegotiation eligibility.
(3) To inform CMS determination of renegotiation eligibility under paragraph (c) of this section, a new indication must be added to the FDA-approved labeling for the selected drug on or before a time specified by CMS and any information regarding off-label use must be voluntarily submitted under § 429.615(b) by the same date.
(d)
Selected drugs for which there has been a material change in any factor listed in section 1194(e) of the Act.
A selected drug with an agreed-upon MFP will be determined to be a renegotiation-eligible drug if there has been a material change in any factor listed in section 1194(e) of the Act.
(1) A change(s) to any factor listed in section 1194(e) of the Act will be considered to be material if the change(s) would reasonably be expected to meaningfully alter CMS' consideration of the factor within the context of renegotiation offers and counteroffers, including the initial offer as described in subpart F of this part, as compared with CMS' consideration of the factor within the context of offers, including the initial offer, and, if applicable, counteroffers, during the most recent prior negotiation or renegotiation for the selected drug.
(2) CMS reviews information available prior to a time specified by CMS pertaining to section 1194(e)(1) or section 1194(e)(2) factors in accordance with § 429.510(f) to determine if there has been a material change.
(3) CMS considers information provided in voluntary submissions described at § 429.615(a), if any, to determine if there has been a material change.
(a)
Selection of renegotiation-eligible drugs due to a change in monopoly status.
All selected drugs determined to be renegotiation-eligible per § 429.605(b) will be selected for renegotiation.
(b)
Selection of renegotiation-eligible drugs due to a new indication or a material change in any factor listed in section 1194(e) of the Act.
Selected drugs determined to be renegotiation-eligible per § 429.605(c) and (d) will be selected for renegotiation if CMS expects renegotiation is likely to result in a significant change to the previously negotiated or renegotiated MFP of such drug.
(1) CMS expects renegotiation is likely to result in a significant change to the MFP if the criteria described in paragraphs (b)(1)(i) and (1)(ii) of this section are both met.
(i) CMS determines the new indication(s) or material change(s) to any factor(s) listed in either section 1194(e)(1) of section 1194(e)(2) of the Act would be likely to result in a renegotiated MFP that represents at least a 15 percent or greater change to the current MFP of the selected drug upon engaging in renegotiation with the Primary Manufacturer.
(A) The percent change may be an increase or a decrease in the MFP.
(ii) CMS determines that a 15 percent or greater change in the MFP following
( printed page 36363)
renegotiation would have a significant impact on the Medicare program.
(2) To determine if the criteria in paragraph (b)(1) of this section are met, CMS conducts a holistic inquiry based on the totality of the information available and the circumstances of the renegotiation-eligible drug.
(i) The scope of information considered may extend beyond the scope of information reviewed for renegotiation eligibility as described at § 429.605(c) and (d) to include consultations with FDA, clinicians, patients, patient organizations, and researchers, a CMS-led review of the information sources described at § 429.510(b)(1), (c)(1), (e), and (f) pertaining to section 1194(e) factors or any other information to support CMS' determination.
Data collection to inform renegotiation eligibility, selection, and renegotiation of the MFP for a selected drug.
(a)
Voluntary information submission from Primary Manufacturers to inform renegotiation eligibility and selection for selected drugs.
A Primary Manufacturer of a selected drug may voluntarily submit information to CMS regarding the addition of a new indication to the selected drug or new information regarding the factors listed in section 1194(e) of the Act to CMS in the form and manner and by the deadline specified by CMS. Information submitted by a Primary Manufacturer will be used to inform renegotiation eligibility, selection, and renegotiation of the MFP for a selected drug.
(b)
Data collection from Primary Manufacturers and other interested parties for renegotiation of the MFP.
After publication of the drugs selected for renegotiation, if any, in accordance with § 429.100(b)(2), in a form and manner to be specified by CMS, and by 11:59 p.m. PST on March 1 of the year of the selected drug publication date for the initial price applicability year for which the drug was most recently selected for renegotiation:
(1)
Primary Manufacturer-required data.
In accordance with the requirements of the Negotiation Program Agreement as set forth in § 429.200, the Primary Manufacturer of a drug selected for renegotiation must submit the following information for such drug for the initial price applicability year for which the drug was selected for renegotiation, inclusive of the NDC-11s of the selected drug manufactured, marketed, controlled, or sold by a Secondary Manufacturer, to CMS in a form and manner specified by CMS:
(i) The information specified at § 429.505(b)(2)(i) through (v); and
(ii) For drugs selected for renegotiation that were selected originally for negotiation for initial price applicability year 2026 or 2027 and have not previously been selected for renegotiation for initial price applicability year 2028 or thereafter, the information specified below for all NDC-11s of the selected drug payable under Part B, not covered under Part D, and for which the Primary Manufacturer did not include such information with the Primary Manufacturer's data submission for the initial price applicability year for which the selected drug was first selected for negotiation:
(A) Non-FAMP, unit type, and total unit volume for each NDC-11, if available, for the same calendar years for which non-FAMP data was reported in the Primary Manufacturer's data submission for the negotiation period in which the selected drug's MFP was negotiated.
(B) When there are at least 30 days of commercial sales data but less than a calendar quarter of data to calculate the non-FAMP in a calendar year, the non-FAMP submitted for that calendar quarter in paragraphs (b)(1)(ii)(A) and (B) of this section should reflect the temporary non-FAMP predicated upon the first 30 days of commercial sales data.
(2) In accordance with the requirements of its Negotiation Program Agreement as set forth in § 429.200, a Primary Manufacturer is required to update the information submitted in accordance with paragraph (b)(1) of this section if the Primary Manufacturer becomes aware that any such information has changed or is otherwise inaccurate.
(3) Interested parties may submit the data specified in § 429.505(d)(3).
(a)
General.
CMS establishes a renegotiation process consistent with the negotiation methodology and process established under section 1194(b) of the Act, to the extent practicable.
(b)
Determining the ceiling for renegotiation.
CMS will not make an offer or agree to a counteroffer for an MFP that exceeds the ceiling specified in section 1194(c) of the Act. Subject to § 429.440(b)(4), CMS calculates the ceiling for drugs selected for renegotiation using the following process:
(1) With respect to selected drugs negotiated for initial price applicability years 2026 and 2027 and have not been renegotiated in initial price applicability year 2028 or thereafter, CMS incorporates NDC-11s that are payable under Part B, if applicable. Specifically, CMS:
(i) For a selected drug that is both covered under Part D and payable under Part B, calculates the payment amount under section 1847A(b)(4) of the Act, as set forth in § 429.425, for the calendar year that is 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation of the previously agreed-upon MFP and the sum of the plan-specific enrollment weighted amounts calculated at the time of the previous negotiation.
(ii) Calculates the average non-FAMP amount(s) used in the calculation set forth in § 429.620(b)(2) using the non-FAMP and Part B utilization data for NDC-11s set forth in § 429.615(b)(1)(i) for the same calendar years for which non-FAMP data was reported during the original negotiation and the non-FAMP and Part D utilization data previously used to calculate the average non-FAMP at the time of the previous negotiation.
(2) CMS updates, as necessary, the applicable percent specified in section 1194(c)(3) of the Act to reflect the monopoly status, which is set forth in § 429.435(a)(4)(vi)(A)(1) through (3), of the drug selected for renegotiation and is determined based on its initial approval date, as set forth in § 429.125(a)(1)(i), and the initial price applicability year for which the drug is selected for renegotiation.
(3) With respect to all selected drugs with an agreed-upon MFP that are renegotiated, CMS, as applicable, updates the amounts considered for the ceiling as set forth in § 429.410, subject to § 429.440(b)(4), for inflation by adjusting for the percent increase in the CPI-U (as defined in § 429.20) from July of the calendar year that is 2 years prior to the initial price applicability year of the most recently published MFP through July of the calendar year prior to the selected drug publication date for the initial price applicability year for which the drug is selected for renegotiation.
(c)
Negotiation factors.
CMS considers the negotiation factors listed at section 1194(e)(1) and (e)(2) of the Act and as set forth in § 429.505 as the basis for determining the offers and counteroffers in the renegotiation process inclusive of information submitted or shared about the factors listed at sections 1194(e)(1) and (e)(2) of the Act in any prior negotiation or renegotiation(s).
(d)
Methodology for developing an initial offer.
CMS follows the process as set forth in § 429.510 to the extent
( printed page 36364)
practicable to develop the initial offer for renegotiation.
(e)
Engagement with Primary Manufacturers and interested parties.
CMS follows the process as set forth in § 429.515 for drugs selected for renegotiation to the extent practicable.
(f)
Provision of CMS' initial offer and concise justification.
CMS follows the process as set forth in § 429.520 to the extent practicable.
(g)
Renegotiation written counteroffers.
The renegotiation written counteroffer follows the process as set forth in § 429.525 to the extent practicable.
(h)
Additional price exchange opportunities.
The additional price exchange opportunities follow the process as set forth in § 429.530 to the extent practicable.
(i)
Notification of final offer and determination that renegotiations have finished.
CMS follows the process as set forth in § 429.535 to the extent practicable.
(j)
Application of the MFP across dosage forms and strengths.
CMS applies the renegotiated MFP across dosage forms and strengths as set forth in § 429.600(b)(2) to the extent practicable.
(k)
Publication of the MFP.
For drugs for which there is agreement on a renegotiated MFP, CMS publishes the renegotiated MFP and related information as set forth in § 429.705 to the extent practicable.
(l)
Establishment of MFPs after the negotiation deadline.
CMS follows the process set forth in § 429.710 in circumstances where a renegotiated MFP is agreed upon after the deadline for the conclusion of negotiations as set forth in § 429.535(b) to the extent practicable.
Application of the MFP across dosage forms and strengths.
(a)
General principles for MFP application.
CMS has the administrative duty to establish procedures to compute and apply the MFP across different dosage forms and strengths of the selected drug and not based on the specific formulation, package size, or package type of such drug.
(1) A single MFP will be applied across NDC-9s and HCPCS codes, as set forth in § 429.100(d), as applicable, for a 30-day equivalent supply and will be used to calculate an MFP per unit for each NDC-9 of the selected drug and an MFP per billing unit for each billing and payment code associated with the selected drug, as applicable.
(b)
Procedures to compute and apply the MFP across dosage forms and strengths.
CMS calculates and applies the MFP across dosage forms and strengths as follows:
(1)
Calculate Annual WAC per Unit.
For each selected drug and calendar quarter of the year that begins 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation, CMS—
(i) For each NDC-11—
(A) Divides the WAC quarterly units by the total WAC annual units (as reported by the Primary Manufacturer under § 429.505(b(2)(v)); and
(B) Multiplies the quotient determined in paragraph (b)(1)(i)(A) of this section by the quarterly WAC per unit for such NDC-11 (as reported by the Primary Manufacturer under § 429.505(b)(2)(v)) to calculate the weighted quarterly WAC per unit.
(ii) For each selected drug, sums the resulting weighted quarterly WAC per unit amounts from paragraph (b)(1)(i)(B) of this section for all NDC-11s to calculate the annual WAC per unit.
(2)
Calculate average number of units per 30-day equivalent supply.
(i) For each NDC-11, CMS calculates the total units dispensed/administered, as reported on PDE data or Part B data for the year that begins 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation.
(A) For NDCs only present in PDE data: total units dispensed/administered are defined as the total quantity dispensed for an NDC-11, summed across the calendar year.
(B) For NDCs only associated with HCPCS codes present in Part B data: total units dispensed or administered are defined as the total billing units sold for an NDC-11 as determined using the unit allocation and standardization methodology described in § 429.425(a)(3), summed across the calendar year.
(C) For NDCs that are both present in PDE data and associated with HCPCS codes present in Part B data: total units dispensed/administered are defined as the sum of the quantity dispensed for the NDC-11 and the total billing units sold for the NDC-11 as determined using the unit allocation and standardization methodology described in § 429.425(a)(3), summed across the calendar year.
(ii) For each NDC-11, CMS divides the total units dispensed/administered, as calculated in paragraph (b)(2)(i) of this section, by the total 30-day equivalent supply, as determined using the 30-day equivalent supply methodology described at § 423.104(d)(2)(iv)(A)(2) for Part D and at § 429.415(a)(2)(v) for Part B, to calculate the average number of units per 30-day equivalent supply.
(3)
Calculate WAC per 30-day equivalent supply ratio.
(i) For each NDC-11, CMS multiplies the annual WAC per unit, calculated as described at paragraph (b)(1) of this section, by the average number of units per 30-day equivalent supply, as described at paragraph (b)(2) of this section, to calculate the WAC per 30-day equivalent day supply for that NDC-11.
(ii) For each NDC-11, CMS divides the total 30-day equivalent supply from paragraph (b)(2)(ii) of this section for such NDC-11 by the total 30-day equivalent supply summed across all applicable NDC-11s within an NDC-9 and then multiplies such quotient by the amount calculated in paragraph (b)(3)(i) of this section to calculate the weighted WAC per 30-day equivalent supply per NDC-11.
(iii) For each NDC-9, CMS sums the resulting amounts from paragraph (b)(3)(ii) of this section across all NDC-11s to calculate the WAC per 30-day equivalent supply for that NDC-9.
(iv) For each NDC-9, CMS divides the total 30-day equivalent supply for that NDC-9 by the total 30-day equivalent supply summed across all NDC-9s and then multiplies this quotient by the resulting amount from paragraph (b)(3)(iii) of this section to calculate the weighted WAC per 30-day equivalent supply per NDC-9.
(v) CMS sums the resulting amounts from paragraph (b)(3)(iv) of this section across all NDC-9s of the selected drug to calculate the WAC per 30-day equivalent supply for the selected drug.
(vi) For each NDC-9, CMS divides the amount from paragraph (b)(3)(iii) of this section by the amount from paragraph (b)(3)(v) of this section to calculate the WAC per 30-day equivalent supply ratio for that NDC-9.
(4)
Calculate NDC-9 MFP per Unit and MFP per Billing Unit.
(i) To determine the NDC-9 MFP per unit, CMS takes the following steps:
(A) For each NDC-9, CMS multiplies the single MFP for the selected drug by the amount resulting from paragraph (b)(3)(vi) of this section to calculate the MFP per 30-day equivalent supply for that NDC-9.
(B) For each NDC-9, CMS divides the amount resulting from paragraph (b)(4)(i) of this section by the quotient of the total number of units dispensed/administered divided by the total 30-day equivalent supply to calculate the NDC-9 MFP per unit at the NCPDP unit (for example, tablet).
( printed page 36365)
(ii) For NDC-11s associated with HCPCS codes present in Part B data, CMS further converts the MFP per NCPDP unit of each NDC-9 into an MFP per billing unit using the following steps:
(A) For each NDC-9, CMS converts the resulting amount from paragraph (b)(4)(i)(B) of this section to an NDC-11 MFP per billing unit, as necessary. First by multiplying NDC-9 MFP per unit calculated in paragraph (b)(4)(i)(B) of this section by the manufacturer submitted total NDC-11 package NCPDP unit quantity, for NDC-11s associated with a HCPCS code, to get an NDC-11 MFP per package. Then, by dividing this product by the HCPCS billing units per package for that NDC-11 to get an NDC-11 MFP per billing unit.
(B) For each NDC-11, CMS sums the units reported by manufacturers in the ASP portal for all quarters for the calendar year that begins 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation for each NDC-11 associated with a given HCPCS code.
(C) For each NDC-11, CMS divides the resulting amount from paragraph (b)(4)(ii)(B) by the sum of units reported by manufacturers in the ASP portal for all quarters for the calendar year that begins 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation for all NDC-11s that share the same HCPCS code and belong to the selected drug.
(D) For each NDC-11, CMS multiplies the resulting amount from paragraph (b)(4)(ii)(A) of this section by the resulting amount from paragraph (b)(4)(ii)(C) of this section.
(E) For each HCPCS code, CMS sums the resulting amount from paragraph (b)(4)(ii)(D) of this section across all NDC-11 associated with the HCPCS code to yield the MFP per billing unit.
(c)
Application of the MFP to NDC-11s and HCPCS codes associated with a new NDA or BLA of the selected drug, new NDC-11s and HCPCS codes for existing NDAs or BLAs of the selected drug, and NDC-11s of the selected drug with insufficient PDE, Part B data, or WAC data for the calendar year that begins 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation.
CMS applies the MFP to NDCs associated with new NDAs or BLAs of the selected drug, new NDC-11s and HCPCS codes for existing NDAs or BLAs of the selected drug, and to NDCs of the selected drug with insufficient PDE, Part B data, or WAC data as follows:
(1)
NDC-11s and HCPCS Codes associated with new NDAs or BLAs of the selected drug.
If the Primary Manufacturer of a selected drug receives approval or licensure for a new NDA or BLA, as applicable, for the same active moiety/active ingredient/antigen component (or in the case of a potential qualifying single source drug identified under § 429.125(b)(4), the distinct combination thereof), as identified as set forth at § 429.125(b)(1) through (4), as the selected drug, CMS—
(i) Uses available information to determine the 30-day equivalent supply and the WAC ratio; and
(ii) Includes the NDCs and HCPCS codes associated with the new NDA or BLA, as set forth in § 429.100(c), as appropriate, on the list of NDCs and HCPCS codes of the selected drug and requires that the MFP apply to such NDCs and HCPCS codes.
(2)
New NDC-11s and HCPCS codes for existing NDAs or BLAs of the selected drug.
If new NDC-11s and HCPCS codes for existing NDAs or BLAs of the selected drug are marketed after the initial calculation of WAC ratios, as set forth in paragraph (b)(3) of this section, CMS—
(i) Uses available information to determine the 30-day equivalent supply and the WAC ratio; and
(ii) Includes the new NDCs and HCPCS codes, as set forth in § 429.100(c), as appropriate, on the list of NDCs and HCPCS codes of the selected drug and requires that the MFP apply to such NDCs and HCPCS codes.
(3)
NDC-11s of the selected drug with insufficient data.
If an NDC-11 (or all NDC-11s assigned to a HCPCS code) that has been included on the list of NDCs of the selected drug under § 429.100(c) lacked sufficient PDE, Part B data, or WAC data for the year that begins 3 years prior to the initial price applicability year with respect to which the selected drug was selected for negotiation, CMS—
(i) Uses available information to determine the 30-day equivalent supply and the WAC ratio; and
(ii) Includes the NDCs and HCPCS codes, as appropriate, on the list of NDCs and HCPCS codes of the selected drug and requires that the MFP apply to such NDCs and HCPCS codes.
(4) To determine the 30-day equivalent supply and the WAC ratio using available information for NDC-11(s) (and all NDC-11s assigned to a HCPCS code) of the selected drug described at paragraphs (c)(1) through (c)(3) of this section, CMS takes the following steps:
(i) Determines whether there is an existing NDC that is comparable to such NDC (or comparable to the NDCs associated with such HCPCS code) with sufficient data for the MFP application calculations to be performed as set forth in paragraph (b) of this section.
(A) If an existing, comparable NDC exists, CMS uses the quotient of total units dispensed or administered to 30-day equivalent supply (adjusted as necessary to reflect dosing or unit type differences between the comparable NDCs and the new NDCs as described at paragraphs (c)(1) and (c)(2) of this section or NDCs with insufficient data as described at paragraph (c)(3) of this section), as described at paragraph (b)(2) of this section, and the WAC ratio, as described at paragraph (b)(3) of this section, that was calculated for the existing, comparable NDC for the application of MFP to the new NDC or NDC that lacks sufficient data.
(B) If a comparable NDC does not exist, CMS imputes the quotient of total units dispensed or administered to 30-day equivalent supply using sources such as the FDA-approved label and other sources associated with the new NDC as described at paragraph (c)(1) or (c)(2) of this section or NDC that lacks sufficient PDE, Part B data, or WAC data as described at paragraph (c)(3) of this section, but uses a WAC ratio of 1.0 to apply the MFP to the new NDC as described at paragraph (c)(1) or (c)(2) of this section or NDC that lacks sufficient PDE, Part B data, or WAC data, but uses a WAC ratio of 1.0 to apply the MFP to the NDC that lacks sufficient PDE, Part B data, or WAC data as described at paragraph (c)(3) of this section.
(1)
When CMS accrues sufficient data for NDCs for which CMS imputed the quotient of total units dispensed or administered to 30-day equivalent supply as described at paragraph (c)(4)(B) of this section, CMS adjusts the MFP application by updating the quotient of total quantity dispensed or administered to 30-day equivalent supply based on observed PDE data and Part B data for existing NDCs (or NDCs associated with such HCPCS code)that lacked sufficient WAC, PDE data, or Part B data to be included in the initial calculation of WAC ratios (described in paragraphs (a) and (b) of this section), and new NDCs (and all NDC-11s assigned to such HCPCS code)launched after the initial calculation of WAC ratios.
(2)
CMS determines that sufficient data was accrued as follows:
(i)
One year has elapsed since the NDCs first appeared in PDE records or the HCPCS codes associated with the NDCs appeared in Part B data.
(ii)
The NDC or HCPCS code has accrued the same number of units
( printed page 36366)
dispensed/administered as the NDC-11 that had the fewest units dispensed/administered at the time that the WAC ratios were originally calculated.
(ii) If a new NDC (or NDC that lacks sufficient PDE, Part B data, or WAC data) is assigned to a HCPCS code for which CMS has already calculated an MFP per billing unit, the MFP per billing unit for that HCPCS code will not change, and the new NDC (or NDC that lacks sufficient PDE, Part B data, or WAC data) will be subject to that existing MFP per billing unit.
(d)
Suggestion of Error.
A Primary Manufacturer that believes in good faith that CMS has made an error in the calculations specified in paragraphs (b) and (c) of this section may submit a Suggestion of Error, as set forth in § 429.445(c) and in a form and manner specified by CMS, as set forth in § 429.445(d).
(a)
Publication.
With respect to an initial price applicability year for which a drug is selected for negotiation or renegotiation, as applicable, CMS publishes by November 30 of the year that is 2 years prior to such initial price applicability year the MFP for each such drug for which CMS and the Primary Manufacturer have reached an agreement on an MFP.
(1) CMS publishes all of the following on the CMS website:
(i) The selected drug.
(ii) The initial price applicability year.
(iii) The MFP file which contains the single MFP for a 30-day equivalent supply of the selected drug, NDC-9 MFP per unit price and HCPCS code dosage price and is updated annually to show the inflation-adjusted MFP for the selected drug as set forth in paragraph (a)(2)(i) of this section.
(iv) Whether an MFP between a Primary Manufacturer and CMS is not agreed upon.
(v) Whether a drug is no longer a selected drug and the reason for that change.
(2) For each selected drug and for each year subsequent to the first initial price applicability year of the price applicability period (unless renegotiation occurs as set forth under subpart G of this part), CMS publishes an updated MFP no later than November 30 of the year that is 2 years prior to such subsequent year.
(i) The updated MFP for each selected drug is equal to the MFP that was published for such drug for the previous year, increased by the annual percentage increase in the CPI-U for the 12-month period ending with the July immediately preceding such November 30.
(3) In the case of a selected drug with respect to an initial price applicability year for which the MFP is agreed upon after the MFPs are published for other selected drugs in accordance with § 429.710, CMS publishes the MFP no later than 30 days after the date such MFP is so agreed upon.
(b)
Explanation for the MFP.
CMS publishes explanations for the MFPs no later than March 1 of the year prior to the initial price applicability year.
(1) The explanation for the MFP of each selected drug, or drug selected for renegotiation, subject to the requirements for treatment of confidential and proprietary information described in proposed § 429.300 includes:
(i) The narrative explanation of the MFP;
(ii) Redacted information regarding the negotiation meetings, as applicable, including exchanges of offers and counteroffers, as applicable; and
(iii) The redacted information submitted by a Primary Manufacturer, as set forth in § 429.505(b)(2) or § 429.615(b)(1), as applicable, and the redacted information submitted by interested parties, as set forth in § 429.505(d)(3) or § 429.615(b)(3), as applicable.
(2) If agreement upon an MFP is not reached for a selected drug, neither an MFP nor an MFP explanation is published. CMS indicates on the CMS website that an MFP has not been agreed upon between the Primary Manufacturer and CMS for the selected drug as set forth in paragraph (a)(1)(iv) of this section.
Establishment of MFPs after the negotiation deadline.
If actions or delays by the Primary Manufacturer delay the negotiation process such that the MFP may be agreed to after the end of the negotiation period as described in § 429.535(b), CMS continues to engage in the negotiation process set forth in subpart F of this part. In accordance with the requirement to have a consistent methodology and process for negotiating MFPs, CMS follows timelines consistent with the negotiation process set forth in subpart F of this part and takes the time to complete the established process so described as appropriate for the selected drug.
(a) If actions or delays by the Primary Manufacturer delay the negotiation process, when CMS initiates or resumes the negotiation process, CMS applies the consistent methodology and process with respect to the selected drug based on its status at the time the negotiation process occurs, including with respect to renegotiation, as applicable, as described in subpart G of this part.
(b) If the manufacturer and CMS have completed each step of the negotiation process as detailed in subpart F of this part, including the issuance of a final offer in accordance with § 429.535(a), and then, after the statutory end of the negotiation period as described in § 429.535(b), the Primary Manufacturer of a selected drug wishes to agree to an MFP, the Primary Manufacturer must notify CMS in writing that it would like to accept the final offer from CMS.
(a)
Monitoring and assessment.
CMS monitors and evaluates Primary Manufacturer compliance with the Negotiation Program Agreement as described in § 429.200, including compliance with all applicable requirements and conditions set forth in sections 1191 through 1198 of the Act and all applicable guidance and regulations, including this part, implementing those provisions and any changes to the Act that affect the Negotiation Program.
(b)
Primary Manufacturer cooperation with CMS compliance monitoring activities.
Primary Manufacturers must cooperate with CMS compliance monitoring activities. In accordance with the Negotiation Program Agreement as set forth in § 429.200, Primary Manufacturers must cooperate with CMS monitoring and evaluation of Primary Manufacturer compliance as specified in subsection (a), including, but not limited to, providing complete and accurate responses to CMS written requests for clarifications, corrections, and additional information, and complying fully with all requests for corrective action, in a form and manner specified by CMS. Such requests will include a date by which the Primary Manufacturer must submit a complete, accurate, and relevant response, or take any other corrective action requested. Non-responsive, incomplete, or inaccurate responses will not be considered compliant. Failure to provide a timely, accurate, and complete response is a violation of the Negotiation Program Agreement as described in § 429.200, and may result in the Primary Manufacturer being subject to a civil monetary penalty as described in § 429.1005.
( printed page 36367)
(c)
Actions to address Primary Manufacturer noncompliance.
If CMS concludes that the Primary Manufacturer is noncompliant with one or more of the requirements of the Negotiation Program Agreement, including all applicable requirements and conditions set forth in sections 1191 through 1198 of the Act and all applicable guidance and regulations, including this part, implementing those provisions and any changes to the Act that affect the Negotiation Program, CMS may take one or more of the following actions:
(1) Provide a written notice to the Primary Manufacturer of the violation(s).
(2) Request the Primary Manufacturer take specific corrective action to address the noncompliance, in a form and manner, and by the deadline specified by CMS.
(3) Impose a civil monetary penalty on the Primary Manufacturer as set forth in subpart K of this part.
(a)
General.
CMS must impose a civil monetary penalty on a Primary Manufacturer that has entered into a Negotiation Program Agreement as set forth in § 429.200, and that fails to comply with a requirement determined by CMS to be necessary for the purposes of administering and monitoring compliance with the Negotiation Program including without limitation the requirement to submit information in accordance with section 1193(a)(4) of the Act.
(b)
Determination of the civil monetary penalty amount.
CMS must impose a civil monetary penalty for such violation as set forth in § 429.1005(a) in the amount of $1,000,000 as adjusted annually under 45 CFR part 102, for each day of such violation as set forth in § 429.1005(a).
(c)
Accrual of civil monetary penalty.
The civil monetary penalty accrues for each day of such violation as set forth in § 429.1005(a), starting on the first day of such violation, until—
(1) The Primary Manufacturer has provided all required information or otherwise taken any corrective action determined by CMS to be necessary to address such violation, including, as applicable, providing documentation to evidence that the Primary Manufacturer has provided all past due information; or
(2) The Negotiation Program Agreement is terminated as described in § 429.205(a).
(d)
Notice.
When a civil monetary penalty is imposed under this section, CMS sends a notice to the Primary Manufacturer in accordance with § 429.1020(a) after the civil monetary penalty stops accruing under § 429.1005(c).
Provision of false information related to the biosimilar delay and Temporary Floor for Small Biotech Drugs.
(a)
General.
CMS must impose a civil monetary penalty as follows:
(1) On a Biosimilar Manufacturer for each item of false information the Biosimilar Manufacturer knowingly provides to CMS for use in applying the aggregation rule described at § 429.110(c)(1)(iv)(A).
(2) On a Primary Manufacturer for each item of false information the Primary Manufacturer knowingly provides to CMS for use in applying the test to determine if a selected drug is eligible for the Temporary Floor for Small Biotech Drugs described at § 429.440(b)(2).
(b)
Determination of the civil monetary penalty amount.
CMS must impose a civil monetary penalty in the amount of $100,000,000 as adjusted annually under 45 CFR part 102, per item of false information described in § 429.1010(a).
(c)
Notice.
When a civil monetary penalty is imposed under this section, CMS sends a notice to the Biosimilar Manufacturer or Primary Manufacturer, as applicable, in accordance with § 429.1020(a).
(a)
General.
CMS must impose a civil monetary penalty on a Reference Manufacturer that fails to pay the rebate amount set forth in § 429.110(i)(4) by the payment deadline as set forth in section § 429.110(i)(2) for such selected drug.
(b)
Determination of the civil monetary penalty amount.
CMS must impose a civil monetary penalty for each violation described at § 429.1015(a) in an amount equal to 10 times the amount of the biosimilar delay rebate as set forth at § 429.110(i)(4) that the Reference Manufacturer failed to pay.
(c)
Notice.
When a civil monetary penalty is imposed under this section, CMS sends a notice to the Reference Manufacturer in accordance with § 429.1020(a).
(a)
Notice of imposition of civil monetary penalties.
If CMS makes a determination to assess a civil monetary penalty described in this subpart K, CMS sends a written notice of its decision to impose a civil monetary penalty to include the following:
(1) A description of the basis for the determination.
(2) The basis for the penalty.
(3) For civil monetary penalties imposed in accordance with § 429.1005, the start date of the penalty accrual.
(4) For civil monetary penalties imposed in accordance with § 429.1005, the end date of the penalty accrual.
(8) Information about where to file the request for a hearing.
(b)
Collection.
(1) A manufacturer must pay the civil monetary penalty in full within 60 calendar days after the date of the notice of imposition of a civil monetary penalty from CMS as set forth in paragraph (a) of this section.
(2) In the event a manufacturer requests a hearing, in accordance with 42 CFR part 423, subpart T, the manufacturer must pay the amount in full within 60 calendar days after the date of a final decision by the Departmental Appeal Board, to uphold, in whole or in part, the civil monetary penalty.
(3) If the 60th calendar day described in paragraphs (b)(1) and (2) of this section is a weekend or a Federal holiday, then the timeframe is extended until the end of the next business day.
(c)
Appeal procedures for civil monetary penalties.
Section 1128A(c)(2) of the Act provides that CMS may not collect a civil monetary penalty until the affected party has had notice and the opportunity for a hearing. Any appeal proceedings will be governed by the provisions of 42 CFR part 423, subpart T.
(1) Primary Manufacturers may appeal the following determinations, subject to the limitations on administrative and judicial review in section 1198 of the Act:
(i) A CMS determination to assess a civil monetary penalty under §§ 429.1000 through 429.1015.
(ii) The calculation of the amount of the civil monetary penalty.
(2) If CMS decides to impose a civil monetary penalty, CMS provides the manufacturer with notice in accordance with the process set forth in paragraph (a) of this section.
(3) A manufacturer has a right to a hearing following a decision by CMS to impose a civil monetary penalty following the administrative appeal process and procedures established in 42 CFR part 423, subpart T.
( printed page 36368)
(d)
Other applicable provisions.
The provisions of section 1128A of the Act (except subsections (a) and (b) of section 1128A of the Act) apply to civil monetary penalties under this section to the same extent that they apply to a civil monetary penalty or procedures under section 1128A of the Act.
(e)
Bankruptcy.
In the event that a manufacturer declares bankruptcy, as described in title 11 of the United States Code, and fails to pay either the biosimilar delay rebate amount owed or the total sum of civil monetary penalties imposed, the government reserves the right to file a proof of claim with the bankruptcy court to recover the unpaid amount of the rebates and civil monetary penalties owed by the manufacturer.
Robert F. Kennedy, Jr.,
Secretary, Department of Health and Human Services.
Footnotes
1.
The most recent final guidance published for the Negotiation Program is the Medicare Drug Price Negotiation Program: Final Guidance, Implementation of Sections 1191—1198 of the Act for Initial Price Applicability Year 2028 and Manufacturer Effectuation of the Maximum Fair Price in 2026, 2027, and 2028, available at:
https://www.cms.gov/files/document/ipay-2028-final-guidance.pdf.
12.
CMS would publish one list with respect to each initial price applicability year. The list would include the selected drug list of the drugs selected for negotiation for the initial price applicability year, as well as drugs selected for renegotiation, if any.
13.
For simplicity, we hereinafter use the term “active moiety/active ingredient” to refer to the active moiety, active ingredient, antigen component, or, in the case of a potential qualifying single source drug identified under the general fixed combination drug policy proposed at § 429.125(b)(4), the distinct combination of active moieties, active ingredients, or antigen components, that we propose to identify as specified at proposed § 429.125(b)(1) through (b)(4). In limited cases, we refer to active moieties, active ingredients, and antigen components in the plural (that is, “active moieties/active ingredients/antigen components” or “active moiety(ies)/active ingredient(s)/antigen component(s)) when we believe such terminology provides greater clarity to the discussion.
18.
This removal would encompass qualifying single source drugs that have been selected for initial price applicability years 2026 and 2027 based on Part D total expenditures.
20.
As described in Negotiation Program Guidance, we provide that, for operational reasons at this time, MFP refunds would not be required for PDE records for selected drugs that were billed as compounds. For alignment, we provide in proposed § 429.120 that PDE records with a compound code indicating the PDE record is for a compounded drug would be excluded from the PDE data used to calculate total expenditures under Part D used for the low-spend Medicare drug exclusion (proposed § 429.125(e)(2)) and to identify negotiation-eligible drugs and selected drugs (proposed §§ 429.115 and 429.105). We are proposing to apply this same exclusion to the ceiling for the MFP (proposed § 429.410), the Net Part D Plan Payment and Beneficiary Liability of a therapeutic alternative(s) (proposed §§ 429.20 and 429.510(d)), and the application of the MFP across dosage forms and strengths (proposed § 429.700). A PDE record for a selected drug billed as a compound refers to a PDE record with a compound code field equal to “2=Compound.” We would only use PDE records with a compound code field equal to “1=Not a Compound.” A Part B claim billed as a compounded drug refers to Part B claims billed with HCPCS code J7999. For consistency with the treatment of compounded drugs covered under Part D, we also would exclude Part B claims billed as compounded drugs when calculating the low-spend Medicare drug exclusion, the identification of negotiation-eligible drugs and selected drugs, the ceiling for the MFP, and the application of the MFP across dosage forms and strengths.
23.
Final action for MA encounter data for Part B items and services indicates the encounter was accepted by CMS and not subsequently voided by the Medicare Advantage organization or superseded by another encounter accepted by CMS.
25.
In the context of identifying qualifying single source drugs and calculating total expenditures for purposes of identifying negotiation-eligible drugs and selected drugs, in this proposed rule we use the term “aggregation” to refer to the process of identifying the formulations and dosage forms and strengths that constitute a qualifying single source drug, and that, if applicable, CMS will use to calculate total expenditures when determining whether such drug is a negotiation-eligible drug or selected drug.
28.
CMS is interpreting the reference to “licensed under section 351(a) of such Act” to mean licensed or deemed licensed under section 351(a) of the PHS Act. Section 351(a) of the PHS Act addresses the licensure of a biological product.
29.
If a drug product or biological product included active moiety/active ingredient/antigen component X, active moiety/active ingredient/antigen component Y, and an additional active moiety/active ingredient/antigen component Z that did not meet the criteria set forth in proposed § 429.125(b)(4)(i), CMS would not include such drug product or biological product under the same potential qualifying single source drug.
30.
The determination of the date of licensure for a potential qualifying single source drug that is a biological product, including a potential qualifying single source drug identified under § 429.125(b)(4)(i), is subject to the proposals at § 429.125(c)(2)(i) and (c)(2)(ii) for a biological product that is a vaccine for infectious diseases, and a biological product with an approved NDA that was deemed to be a BLA.
31.
For a biological product with an approved application under section 505(c) of the FD&C Act that was deemed to be a BLA under section 7002(e)(4)(B) of the BPCI Act, as amended by the Further Consolidated Appropriations Act of 2020, we would consider the approval date determined in accordance with section 7002(e)(4)(B) of the BPCI Act to be the licensure date for purposes of identifying the time since licensure under section 1192(e)(1)(B)(ii) of the Act.
32.
For orphan drug designations withdrawn after August 12, 2013, the FDA Orphan Drug Product designation database includes the date of such withdrawal. For orphan drug designations withdrawn prior to or on August 12, 2013, we would use August 12, 2013, as the date on which the orphan designation is withdrawn for purposes of identifying the first day after the drug or biological product's approval or licensure that such drug or biological product does not qualify for the orphan drug exclusion.
33.
For purposes of applying the orphan drug exclusion, CMS understands “approved indication,” as that term is used in section 1192(e)(3)(A) of the Act, to refer to the FDA-approved indication that is described in information included in drug labeling per 21 CFR 201.57(c)(2) or other applicable FDA regulation(s).
34.
For purposes of the Negotiation Program, we use the term “withdrawn” with respect to an orphan drug designation to refer to a voluntary withdrawal of an orphan drug designation as described at 23 CFR 316.24 or a revoked orphan drug designation as described at 23 CFR 316.29.
43.
CMS is interpreting the reference to “licensed under section 351(a) of such Act” to mean licensed or deemed licensed under section 351(a) of the PHS Act. Section 351(a) of the PHS Act addresses the licensure of a biological product.
46.
The CGDP, established under section 1860D-14A of the Act, remained in place through December 31, 2024. Because the IRA sunset the CGDP effective December 31, 2024 and all manufacturer CGDP agreements automatically terminated as of such date, CMS has removed references to the CGDP in discussion of Primary Manufacturer termination. CGDP requirements are codified in subpart W of 42 CFR part 423.
49.
When discussing a selected drug that is payable under Part B but is not paid according to section 1847A(b)(4) of the Act in § 429.410(b)(1)(iv) and (v), an example would be a selected drug which is paid on the basis of 95 percent of Average Wholesale Price (AWP).
50.
CMS did not use the term “selected drug” in this proposal because there could be a shared HCPCS code or different NDA/BLA holders, especially for therapeutic alternative(s) of the selected drug.
51.
Employer sponsored plans that receive the retiree drug subsidy and health plans that offer creditable prescription drug coverage would not be included because they are not Part D plans.
52.
In contrast, for selected drugs not covered under Part D but which are payable under Part B and not paid according to section 1847A(b)(4) of the Act, we do not believe any of the amounts described in section 1194(c)(1)(B) of the Act are applicable, so we propose that section 1194(c)(1) of the Act is best read as permitting the agency to apply only the amount described under section 1194(c)(1)(C) of the Act for such drugs.
53.
Section 1192(d) of the Act implemented the SBE for initial price applicability years 2026, 2027, and 2028, which included criteria for determining if a qualifying single source drug was a Small Biotech Drug. Section 1194(d) of the Act directs CMS to use the same criteria when determining whether a selected drug is a Small Biotech Drug. Section 1194(f)(4)(B) extends the Temporary Floor for Small Biotech Drug provisions to certain drugs selected for renegotiation. Therefore, when discussing the criteria to determine a Small Biotech Drug to determine the eligibility of the Temporary Floor for Small Biotech Drugs, we use the term “selected drug” as defined in proposed § 429.20 and “drug selected for renegotiation” in lieu of “qualifying single source drug”.
55.
For purposes of the Temporary Floor for Small Biotech Drugs and implementing section 1192(d)(2)(B)(ii) of the Act, to determine whether the acquiring entity meets the definition of a specified manufacturer under section 1860D-14C(g)(4)(B)(ii) of the Act, CMS will use the determination made by CMS under the Manufacturer Discount Program as to whether the acquiring entity is a “specified manufacturer.” The Part D Manufacturer Discount Program ICR (CMS-10846, OMB control no. 0938-1451) is available for viewing at
https://www.reginfo.gov/public/do/PRAViewICR?ref_nbr=202307-0938-003
(select “all” to see full details).
59.
For biological products with approved applications under section 505 of the FD&C Act as of March 23, 2020, that were deemed to be approved BLAs under section 351 of the PHS Act, effective March 23, 2020, under section 7002(e)(4)(A) of BPCI Act, and that are currently licensed and marketed under section 351 of the PHS Act, CMS will consider March 23, 2020 to be the licensure date for purposes of identifying the time since licensure under section 1192(e)(1)(B) of the Act.
60.
As we propose to codify in § 429.20, section 1194(c)(4)(B)(ii) of the Act expressly excludes a selected drug for which a Primary Manufacturer has entered into a Negotiation Program Agreement with CMS with respect to an initial price applicability year that is before 2030 from the definition of an “extended-monopoly drug”. Therefore, the proposal at § 429.605(b)(1) would apply to drugs selected for negotiation in initial price applicability year 2030 or later.
61.
For biological products with approved applications under section 505 of the FD&C Act as of March 23, 2020, that were deemed to be approved BLAs under section 351 of the PHS Act, effective March 23, 2020, under section 7002(e)(4)(A) of BPCI Act, and that are currently licensed and marketed under section 351 of the PHS Act, CMS will consider March 23, 2020 to be the licensure date for purposes of identifying the time since licensure under section 1192(e)(1)(B) of the Act.
62.
Such additions to the FDA approved labeling include new indications approved in both new NDAs/BLAs and supplements to previously approved NDAs/BLAs.
64.
The Maximum Fair Price Layout file would display the NDC-9 MFP per-unit price to six decimal places. Publishing an NDC-9 MFP per-unit price rounded to the sixth decimal point place aligns with how CMS publishes other prices. Furthermore, publishing an NDC-9 MFP per-unit price rounded to the sixth decimal place would also result in the same agreed-upon MFP per 30-day equivalent when reversing the application of the single MFP across dosage forms and strengths calculations.
71.
As noted in section 90 of the Final CY 2026 Part D Redesign Program Instructions, the section 1860D-4(b)(3)(I)(ii) exception to the IRA's formulary inclusion requirement for selected drugs addresses when a Part D plan sponsor can remove a selected drug from a formulary. Accordingly, this section of the proposed rule applies specifically to negative formulary changes that result in the removal of a selected drug from a formulary. This section does not affect Part D plan sponsors' ability to implement negative formulary changes other than removal where a Part D plan sponsor would continue to include a selected drug on its formulary as required by section 1860D-4(b)(3)(I)(i) of the Act (for example, moving the selected drug to a higher cost-sharing tier or adding utilization management practices) if all applicable requirements are met.
74.
Medicare Drug Price Negotiation Program: Final Guidance, Implementation of Sections 1191-1198 of the Social Security Act for Initial Price Applicability Year 2028 and Manufacturer Effectuation of the Maximum Fair Price in 2026, 2027, and 2028, Section 40.4.
https://www.cms.gov/files/document/ipay-2028-final-guidance.pdf.
77.
See May 2025 All data (XLSX) National Industry-Specific Occupational Employment and Wage Estimates, NAICS 325400—Pharmaceutical and Medicine Manufacturing. Available at:
https://www.bls.gov/oes/tables.htm.
78.
Industry-specific wage estimate not available, see May 2025 All data (XLSX) National Industry-Specific Occupational Employment and Wage Estimates, NAICS 000000—Cross-industry. Available at:
https://www.bls.gov/oes/tables.htm.
79.
Industry-specific wage estimate not available, see May 2025 All data (XLSX) National Industry-Specific Occupational Employment and Wage Estimates, NAICS 000000—Cross-industry. Available at:
https://www.bls.gov/oes/tables.htm.
80.
Industry-specific wage estimate not available, see May 2025 All data (XLSX) National Industry-Specific Occupational Employment and Wage Estimates, NAICS 000000—Cross-industry. Available at:
https://www.bls.gov/oes/tables.htm.
81.
As clarified in the CY 2026 Physician Fee Schedule Final Rule, the average sales price (ASP) reported by a manufacturer must include the MFP, and the ASP is typically the basis for calculating the Part B inflation rebate. (In contrast, the average manufacturer price [AMP] that is the basis for calculating the Part D inflation rebate excludes the MFP per section 1927(k)(1)(B)(i)(VI) of the Act.)
85.
Data based on number of employees listed within the most recently publicly available Annual or Fourth Quarter Report for each Primary Manufacturer during the associated Initial Price Applicability Year at the time this NPRM was drafted.
Use this for formal legal and research references to the published document.
91 FR 36236
Web Citation
Suggested Web Citation
Use this when citing the archival web version of the document.
“Medicare Drug Price Negotiation Program and Medicare Prescription Drug Benefit Program,” thefederalregister.org (June 16, 2026), https://thefederalregister.org/documents/2026-12059/medicare-drug-price-negotiation-program-and-medicare-prescription-drug-benefit-program.